Retinoblastoma treatment requires significant multidisciplinary input, but early detection through raising awareness remains key to improving outcomes. In the second article of a two-part series, Manoj Parulekar discusses retinoblastoma management, screening and research.
This article has been published in two parts. The first section addressed the clinical features, diagnosis and genetic aspects of retinoblastoma. In this article, we will discuss the current management of retinoblastoma patients, screening and surveillance of family members, long-term follow-up and potential future developments.
Treatment of retinoblastoma
Retinoblastoma has evolved from a deadly childhood cancer to a largely curable cancer within the past 40 years. Current treatment strategies aim to salvage the eye and provide the best visual outcome possible. This requires significant multidisciplinary input and should be coordinated by a specialised centre. The various modalities of treatment are discussed below.
Laser treatment: Laser treatment is suitable for primary treatment of smaller tumours, or larger tumours after they have been shrunk to a treatable size with chemotherapy (chemoreduction). Laser treatment is, however, not effective for vitreous seeds (Figure 1).

Figure 1: Tumours before (above) and after treatment with chemotherapy and laser (below).
Laser is delivered through dilated pupils using the indirect ophthalmoscope or microscope. The two common laser wavelengths are 532nM green light and 810nM infrared light. Large spot 810nM treatment heats the tumour slowly (thermotherapy) to produce necrosis and is preferred by many centres [1].
Cryotherapy: Using a special probe applied through the sclera to produce temperatures as low as -60 to -80ºC results in cryonecrosis of tumours. This treatment is suitable for larger, peripheral tumours, or localised vitreous disease close to the retina.
Radiotherapy: This could be in the form of external beam radiotherapy (EBRT, teletherapy) or using a radioactive plaque (brachytherapy). Once the mainstay of treatment, EBRT is very rarely used nowadays due to significant risks of induced secondary malignancies (major risk in germ line cases) within the radiation field, cataracts, dry eyes and soft tissue and bony atrophy. EBRT is reserved for recurrent disease in the only remaining eye, not responsive to all other forms of treatment. Proton beam radiotherapy is used in preference to conventional (photon) radiotherapy where possible to minimise collateral damage.
In contrast, plaque brachytherapy involves attaching a radioactive plaque of Iodine 131I or Ruthenium 106Ru onto the sclera for a specified period (hours to days) to deliver a high dose of radiation to a localised area without the risks of EBRT. It is highly effective against localised vitreous disease, recurrent disease and for elevated tumours where laser is ineffective [2].
Chemotherapy: Retinoblastoma tumours are very sensitive to chemotherapy.
Systemic chemotherapy has been in use for over two decades [3] and is used to shrink the tumours to a size where laser treatment can be effective (chemoreduction) [4]. It is also effective against solid tumours, as well as vitreous and subretinal disease. The most common regime involves carboplatin, etoposide and vincristine (referred to as JOE or CEV chemotherapy) given systemically, delivered through a central line, and typically involves four to six cycles at three to four weekly intervals.
There can be short and long-term side effects of chemotherapy, including high frequency hearing loss with carboplatin and nephrotoxicity, but is generally well tolerated in children. Periocular injections of carboplatin, and topotecan have been used along with systemic chemotherapy to try and boost the intraocular levels of the drug by trans-scleral diffusion to enhance efficacy. The results have been disappointing, and these techniques are rarely used nowadays.
Second line chemotherapy may be used in some cases of recurrent disease after primary chemotherapy – agents used are topotecan, vincristine and doxorubicin.
High dose chemotherapy is used for extraocular disease or metastatic disease, and carries very high morbidity.
Enucleation: This is the oldest treatment, and is curative for intraocular retinoblastoma. It is the treatment of choice for advanced uniocular disease or the worse eye of bilateral cases [5].
The eye is removed with a long segment of optic nerve and sent for histology, and tumour DNA studies to identify the mutations. These mutations are then tested against peripheral blood to establish if it is a germ line or somatic case.
The histopathologist will look for high risk features (invasion of the tumour into the optic nerve beyond the lamina cribrosa, or >3mm invasion of the choroid). If these features are present, systemic chemotherapy will be needed to treat for the risk of potential metastases.
It is usual to replace the lost volume of the enucleated globe with a spherical implant of appropriate size depending on the age of the child. The implant material may be porous such as porous polyethylene (Medpor) or hydroxyapatite, or bioceramic, or non-porous such as silicone or acrylic. A prosthetic shell painted to match the other eye is fitted in due course for cosmesis.
Newer treatment modalities
The most significant advance in the past decade has been new techniques to deliver higher doses of chemotherapeutic drugs into the eye, maximising efficacy, with minimal systemic toxicity.
These include:
- The interventional radiological technique of intra-arterial chemotherapy (IAC), also referred to as selective ophthalmic artery chemotherapy (OAC). This involves a trans-femoral approach to catheterise the ophthalmic artery, allowing delivery of a high dose of single, double or triple agent chemotherapy into the ophthalmic artery. Commonly used drugs are melphalan, topotecan and carboplatin. The commonest use of this treatment is for recurrent or residual disease, especially solid retinal or subretinal disease. It is less effective against vitreous disease. Some centres including ours are now using IAC as first line treatment in selected cases, e.g. for moderate sized tumours in unilateral cases, where the visual prognosis for the eye is poor, and the aim is to avoid enucleation and systemic chemotherapy. This treatment requires skilled interventional neuro-radiology set-up and is expensive.
- Direct injection of drugs into the vitreous cavity intravitreal chemotherapy (IVC).
The long held principle of avoiding entering the eye was challenged with the introduction of this new treatment in 2009, and has been a paradigm shift in management. Vitreous disease is less responsive to all other forms of treatment, and has been a major cause of failure in the past. IVC involves pars-plana injection of one or two chemotherapeutic agents (typically melphalan, topotecan, or carboplatin) directly into the vitreous, after anterior chamber paracentesis, with cryotherapy to the injection site. This highly effective treatment is inexpensive, and does not require a specialised set-up. It can therefore be offered in most retinoblastoma centres in developing countries, which has reduced enucleation rates markedly in recent years (Figure 2).
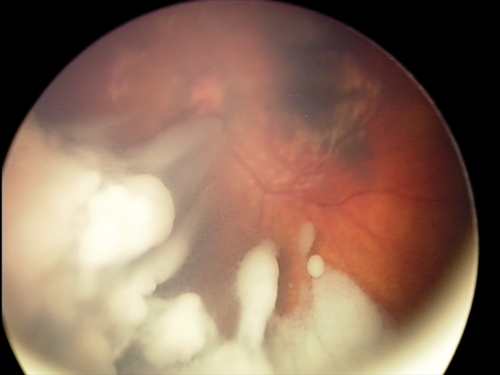
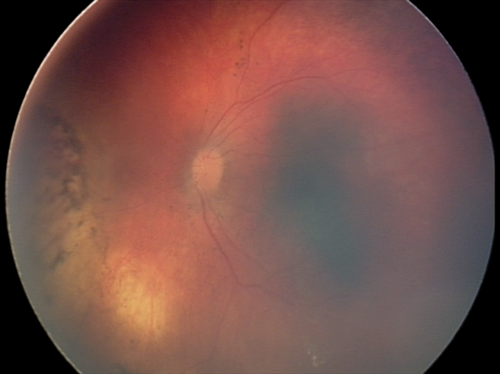
Figure 2. Successful treatment of extensive vitreous disease with intravitreal chemotherapy.
Research directions
The relatively small numbers of cases seen by each centre, lack of a satisfactory animal model, and numerous non-comparable staging systems have hindered retinoblastoma research in the past. A more collaborative approach with multicentre studies currently underway will pave the way for a more evidence based approach to retinoblastoma management.
Current research is directed towards:
- A better understanding of genotype-phenotype relationships in retinoblastoma that will be useful in the multidisciplinary management of this disease.
- A better understanding of pharmacokinetics, and development of less toxic chemotherapy agents for ocular use.
- Newer biological treatments including oncolytic viruses.
- Developing better animal models.
It is likely future research will be directed towards targeted molecular therapy to individualise treatment, and gene therapy to prevent tumour formation.
Treatment principles
Retinoblastoma is a unique cancer by virtue of its confinement within the scleral envelope, and has a 98% cure rate with appropriate treatment. The diagnosis is clinical, and it is important to avoid breaching this envelope with an intraocular procedure such as diagnostic biopsy to avoid the mortality associated with extraocular spread.
A combination of treatment modalities, e.g. chemotherapy with laser, cryotherapy or plaque brachytherapy, helps minimise adverse effects [4,5].
Close monitoring with examinations under anaesthesia (EUA) at decreasing frequency as the child grows older is important for early detection of recurrent or new tumours, with examinations without anaesthesia for older children.
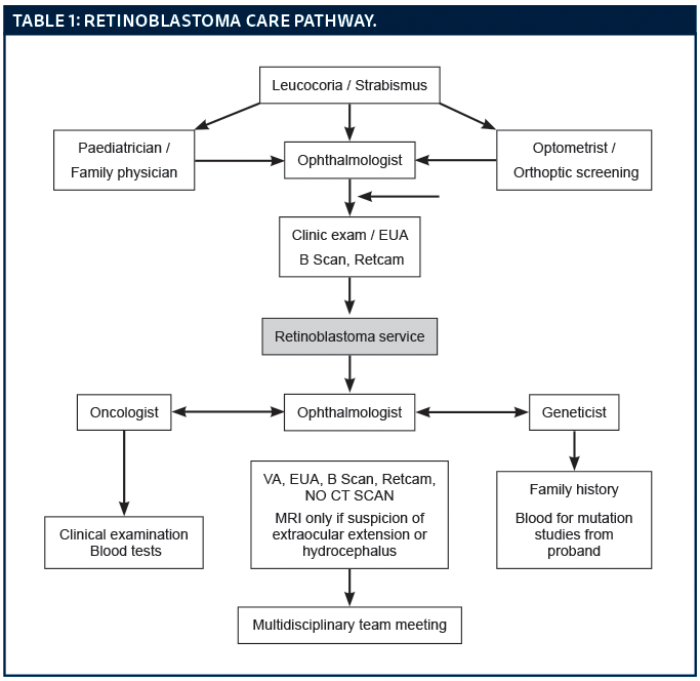
During active treatment, systemic chemotherapy is given over four to six cycles at three weekly intervals, with EUAs before each cycle to monitor response and apply local treatment (laser or cryotherapy). Intra-arterial chemotherapy is typically given every three weeks, over two to four cycles. Intravitreal chemotherapy is given every seven to fourteen days, for three to six cycles. Local treatment may be continued at further EUAs until all tumours are inactive. A typical care pathway is shown in Table 1.
Supportive treatment
- Prosthesis fitting for enucleated eyes – an important part of rehabilitation, usually a few weeks after surgery.
- Psychological support for children and families – to deal with loss of eye, vision and a chronic illness.
- Protective eye wear for the better / remaining eye during contact sport.
- Long-term oncological surveillance especially for germ line cases – this is best undertaken by oncologists.
- Counselling – parents should be counselled soon after diagnosis, and the patients when they reach adolescence about the inheritance of retinoblastoma, and risk to siblings and offspring. The risk of secondary malignancies, advice about risk factors like smoking and how to look out for early warning signs should be discussed with adolescent patients.
- Cataract surgery, if needed, should be delayed for at least one to two years after active treatment.
Prenatal diagnosis – the role of imaging and tissue sampling
If there is a family history of retinoblastoma and the mutation in the affected parent is known, there are several options to prevent retinoblastoma or enable early detection:
- Pre-implantation genetic diagnosis (PIGD) involves screening embryos at the blastocyst stage. One cell is removed from the embryo to look for the mutation. Unaffected embryos are selectively implanted ensuring the foetus is born free of the retinoblastoma mutation and does not require screening. Additionally, there is no risk of second cancers, and no risk to future generations. The obvious disadvantage of this technique is the need for in-vitro fertilisation (IVF) to produce and then select embryos.
- Chorion villous sampling (CVS) or amniocentesis are interventional techniques to obtain fetal tissue samples prenatally and test for the RB mutation [9].
- Prenatal ultrasound – In cases with a family history, and particularly if the child is shown to carry the mutation on CVS or amniocentesis, B scan ultrasonography may be advisable in the later stages of pregnancy to look for development of tumours. Although the sensitivity of this technique is quite low, in experienced hands, it may be possible to detect large tumours developing in the last few weeks of pregnancy. Some centres advocate induction of labour at 36 weeks to enable treatment in such cases. There can, however, be some difficulty giving chemotherapy to preterm babies, and treatment may have to be delayed until term. There are also increased risks of surgical intervention such as caesarean section and forceps in induced births. As a result, there is no consensus and much controversy on this subject.
- Cord blood testing. The obstetric team can collect cord blood and send it to a reference laboratory to test for the Rb mutation.
- Free fetal DNA testing. This exciting new technique involves testing free (extracellular) fetal DNA, which is known to cross the placental barrier. In cases where the mother is healthy and the father is the mutation carrier, maternal blood is tested using DNA amplification. If the mutation is found in maternal blood, one can deduce that it has come from the fetus, which must be carrying the mutation. This can then be confirmed at birth with cord blood testing.
Screening for retinoblastoma
Screening close relatives of retinoblastoma patients is invaluable in early detection and treatment, saving eyes and lives. If the mutation for the index case is known, testing can be offered to relatives to determine if they are at risk of suffering / passing on the disease.
Screening is offered if the mutation is positive or if the mutation is not known for the index case, and risk cannot be excluded.

Most centres will have protocols for screening. A suggested protocol is shown in Table 2. Screening is not needed if the relative does not carry the mutation. This approach helps avoid unnecessary screening, and focuses resources. Ideally the first screening exam for neonates with a close family history of retinoblastoma is performed in the obstetric unit before discharge, and subsequent examinations can be arranged with ophthalmic units or units familiar with retinoblastoma management.
Prognosis
Prognosis for life
Most untreated tumours proceed to local invasion and metastasis to cause death within two years. Occasionally, however, the tumour may spontaneously stop growing to form a retinoma, or necrose to cause phthisis bulbi (shrunken globe).
Most small / medium tumours without vitreous seeding can be successfully treated while preserving useful vision. Overall there is a 98% survival rate (in the developed world). Poor prognostic factors include: size of tumour, optic nerve involvement, extraocular spread and older age at presentation.
Prognosis for vision
The prognosis for vision in retinoblastoma survivors is good, with better than 6/12 vision in the better seeing / remaining eye in over 80% of cases (personal data).
Recurrence
Recurrence can develop within the eye in previously treated tumours, and regular follow-up examinations are essential. Delayed extraocular recurrence in the orbit or distant metastases may be seen in a small number of advanced cases that appeared to be restricted to the eye at initial presentation, and / or were negative on metastatic workup. Metastases may occur several decades after the initial presentation. The management of these cases can be challenging.
Risk of second cancers and the role of long-term surveillance
Patients with germ-line mutations are at increased risk of developing secondary malignancies such as pinealblastoma (trilateral retinoblastoma) and osteogenic or soft tissue sarcomas, melanoma and bladder cancer.
The cumulative risk of second cancers has been reported at 20-48% over 50 years in various studies [10]. This risk is increased with radiation exposure. The role of long-term screening of retinoblastoma survivors is not clearly defined in the literature. Patient education and health awareness play a key role in minimising delay in diagnosis and treatment of second malignancies in these patients.
Summary
There have been significant advances in our understanding and management of retinoblastoma over the past few decades. Early detection to minimise visual morbidity remains the focus of research in countries with universal access to healthcare. Late presentation with life-threatening disease is a major challenge in economically underprivileged countries, and strategies must focus on raising awareness and acceptance of modern treatment methods through health workers. Developing a global network of retinoblastoma treatment centres that share information and offer advice, and global funding initiatives will save lives and vision.
References
1. Shields JA. The expanding role of laser photocoagulation for intraocular tumours. The 1993 H. Christian Zweng Memorial Lecture. Retina 94;14(4):310-22.
2. Abouzeid H, Moeckli R, Gaillard MC, et al. Ruthenium brachytherapy for retinoblastoma. Int J Radiat Oncol Biol Phys 2008;71(3):821-8.
3. Chan HS, Gallie BL, Munier FL, Beck Popovic M. Chemotherapy for retinoblastoma. Ophthalmol Clin North Am 2005;18(1):55-63.
4. Levy C, Doz F, Quintana E, et al. Role of chemotherapy alone or in combination with hyperthermia in the primary treatment of intraocular retinoblastoma:preliminary results. Br J Ophthalmol 1998;82(10):1154-8.
5. Shields CL, Shields JA. Retinoblastoma management: advances in enucleation, intravenous chemoreduction, and intra-arterial chemotherapy. Curr Opin Ophthalmol 2010;21(3):203-12.
6. Abramson DH, Shields CL, Munier FL, Chantada GL. Treatment of retinoblastoma in 2015: agreement and disagreement. JAMA Ophthalmol 2015;133(11):1341-7.
7. Abramson DH, Dunkel IJ, Brodie SE, et al. A phase I/II study of direct intraarterial (ophthalmic artery) chemotherapy with melphalan for intraocular retinoblastoma initial results. Ophthalmology 2008;115(8):1398-404.
8. Munier FL, Gaillard MC, Balmer A, et al. Intravitreal chemotherapy for vitreous disease in retinoblastoma revisited: from prohibition to conditional indications. Br J Ophthalmol 2012;96(8):1078-83.
9. Singh AD, Black SH, Shields CL, Shields JA. Prenatal diagnosis of retinoblastoma. J Pediatr Ophthalmol Strabismus 2003;40(4):222-4.
10. MacCarthy A, Bayne AM, Draper GJ, et al. Non-ocular tumours following retinoblastoma in Great Britain 1951 to 2004. Br J Ophthalmol 2009;93(9):1159-62.
Declaration of competing interests: None declared.
See here for Part 1 of this topic.
COMMENTS ARE WELCOME








