The first of a two part series, this article will discuss the clinical features, diagnosis and genetic aspects of retinoblastoma. Manoj Parulekar is based at Birmingham Children’s Hospital, one of the two designated national retinoblastoma and paediatric ocular oncology treatment centres in the UK.

Figure 1: Advanced retinoblastoma with fungating orbital mass.
Retinoblastoma is a malignant tumour arising from the developing retina. It typically presents in the first two to three years of life, often during infancy [1]. The tumour is confined to the eye in the early stages, and cure rates for intraocular retinoblastoma can be as high as 99%. Extraocular spread (Figure 1) particularly optic nerve and central nervous system involvement carries a very poor prognosis [2], with cure rates below 10%. Early diagnosis and prompt treatment is therefore crucial to save life and vision.
Epidemiology
Retinoblastoma is the commonest primary malignant intraocular tumour of childhood, accounting for up to 1% of all tumours in infancy. The age adjusted annual incidence is 12 per million population [3] under the age of four years.
The incidence of sporadic retinoblastoma is one in 15,000-20,000 live births, with no gender or racial predilection. As expected, the burden of disease is highest in populous developing countries in proportion to the birth rate [3]. The median age at presentation is under 12 months in heritable cases, and closer to 24 months in sporadic cases [4]. Presentation after the age of six years is extremely rare, although there are isolated reports of cases presenting as late as 26 years.
Genetics
Understanding the genetics of retinoblastoma is important when planning management. The tumour arises from primitive cells of the developing retina with loss of function of the Rb tumour suppressor gene (Ch13q14) [4]. The retinoblastoma gene was the first oncogene to be cloned, and is the prototype genetic cancer syndrome. Other genetic cancer syndromes include the Li- Fraumeni syndrome and Xeroderma pigmentosum.
There are two copies (alleles) of the Rb gene in every cell in the body, and at least one functioning allele is required to prevent the development of retinoblastoma in a retinal cell. Loss of both alleles will result in retinoblastoma tumours. Interestingly, in 1973 Knudson, a statistician, proposed that two separate mutational events - M1 and M2 that result in loss or inactivation of both Rb gene copies are required to initiate the tumour on the basis of his epidemiological studies, and this came to be known as the Knudson ‘two-hit’ hypothesis which has stood the test of time [5]. Recent evidence suggests that tumour initiation (retinoma) can occur with two hits, but subsequent mutations (M3-Mn) are necessary for the tumour to grow into retinoblastoma [6]. Loss of segments of the chromosome carrying the Rb1 genetic material, e.g. chromosome 13q14 deletion, can also result in retinoblastoma, and is often associated with developmental delay and dysmorphic features.
The two hits can occur in one of two situations. In the genetic (also referred to as germ-line or heritable) form, every cell in the body is missing one copy of the Rb gene, the mutation occurring at the zygote stage. Every retinal cell is missing one copy of the gene, and can potentially give rise to a tumour. The other situation is the somatic (non heritable) type where a single developing retinal cell loses one copy of the Rb gene during retinal development – the rest of the body cells are normal. This constitutes the first hit. The second hit is a random event, and gives rise to a tumour. This makes it easier to understand why genetic cases often have multiple tumours in one or both eyes (bilateral or unilateral multifocal), while somatic cases are always unilateral and unifocal.
More recently, a new subset of early onset retinoblastoma tumours have been described that do not carry RB1 mutations, but instead carry n-Myc mutation, and are poorly responsive to chemotherapy.
Inheritance patterns
Retinoblastoma may be inherited or occur sporadically. Over 90% of cases are sporadic (with no family history). In most of these cases the mutation is somatic and gives rise to isolated unilateral disease. A third of the sporadic cases arise from new germ-line mutations which are heritable (can be passed to offspring) but not inherited from the parent).
Forty percent of all cases are bilateral (and necessarily germ-line), and 60% are unilateral (which could be somatic or germ-line). Of the unilateral cases, 15% are germ-line, and carry the same risks as bilateral germ-line cases. Germ-line mutations are highly penetrant. Over 90% of children carrying the Rb gene defect will develop retinoblastoma.
It has been suggested that there is an increased incidence of retinoblastoma in babies born of assisted conception. There is, however, insufficient evidence in the literature to support or refute this theory.
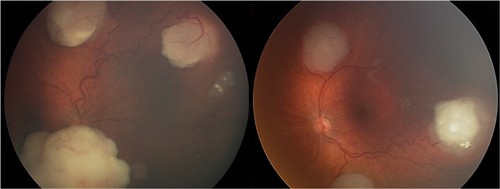
Figure 2: Multiple tumours in both eyes in a four month old with germ-line retinoblastoma.
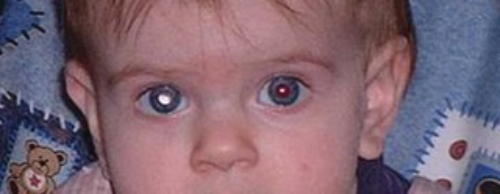
Figure 3: White reflex on digital photography – leukcocoria.
Presentation
Unilateral and bilateral cases differ in the manner and timing of presentation. Bilateral cases often present with poor vision, and nystagmus or searching eye movements as a result of the tumour involving the central part of the retina (macula) in both eyes (Figure 2), or leukocoria (white reflex in the pupil) noticed by the parents in one or both eyes (Figure 3). Early presentation within the first few months of life is usual for bilateral cases, and even at birth in some cases.
Unilateral cases (most of which are somatic mutations) usually present later, often at the age of two to three years or older. However, approximately 15% of unilateral cases carry the germ line mutation, and such cases can present early, often in infancy. The commonest presenting features in unilateral cases are leukocoria (60%) and squint (20%) [7]. Less common modes of presentation are reduced vision detected at school screening, acute red eye, orbital inflammation and excess watering.
If a child presents with unilateral retinoblastoma early in life, it is more likely to be germ-line (heritable) as described earlier, with a high risk of developing more tumours in the same or fellow eye [1]. Careful screening of the fellow eye is essential in all such cases through childhood.
Advanced cases are rarely seen in economically advanced countries. This is more common with unilateral cases. Anterior segment involvement may result in raised pressure in the eye (glaucoma), enlargement of the globe or change in iris colour (heterochromia). Less common presenting features include phthisis bulbi (shrinkage of the globe), pseudohypopyon (cells in the anterior chamber appearing as a layered white material) rubeosis (dilatation of iris vessels) and hyphaema (bleeding into the anterior chamber). Some cases may present with periocular inflammation.
Late presentation with proptosis (forward displacement of the eye) or fungating orbital mass is not unusual in countries with limited access to healthcare and carries a very poor prognosis (Figure 1).
In advanced cases, the tumour can spread along the optic nerve to involve the central nervous system, or through the choroid resulting in haematogenous spread to bones, lungs and abdominal solid organs.
Digital flash photography can highlight the white reflex (Figure 3) in some cases, and lead to earlier detection, and this continues to be the subject of campaigns to raise awareness. Only a minority of cases with a white reflex on photographs will turn out to have retinoblastoma, however. Squint, refractive errors and photographs taken from an angle (off axis) can similarly produce a white reflex, diminishing the specificity of this technique as a screening test. However, the Honduran retinoblastoma campaign was associated with a reduction in the proportion presenting with advanced disease [8].
Diagnosis
The diagnosis of retinoblastoma is essentially clinical, and may be supported by imaging in some cases. Diagnostic biopsy is contraindicated because of the risk of extraocular spread [9].
The typical findings are one or more round white retinal mass(es) growing into the vitreous cavity (endophytic) or growing into the subretinal space (exophytic). Fragments of the tumour can spread as vitreous or sub-retinal seeds (Figure 2).
A characteristic feature highly supportive of the diagnosis is visible calcification within the tumour on ophthalmoscopy. Calcification is also detectable on ultrasonography.
The diagnosis is confirmed histologically if the eye is removed surgically (enucleation – see below). Characteristic histological features include abnormal patterns of retinoblasts such as the Flexner-Wintersteiner rosettes, Homer Wright rosettes, and fleurettes. The histopathologist will also look for features of invasion into the optic nerve or choroid, which would indicate the need for post-enucleation chemotherapy.
“Retinoblastoma is the commonest primary malignant intraocular tumour of childhood, accounting for up to 1% of all tumours in infancy.”
Investigations
Imaging
B scan ultrasonography: confirms the presence of masses in the posterior segment of the eye. Characteristic findings are intra-lesional calcification with high internal reflectivity and acoustic shadowing.
The role of routine MRI scans is still under study. Some centres routinely obtain MRI scans, while others use it if there is suspicion of extraocular (particularly intracranial) spread, if the child presents with signs of raised intracranial pressure (to look for pinealblastoma – trilateral retinoblastoma) or if the diagnosis is in doubt. X-ray and CT scan are avoided as any dose of radiation magnifies the risk of secondary malignancies in germ line cases [10].
Genetic testing
Mutation testing is an essential investigation, and can be performed on peripheral blood and tumour tissue if available (from the enucleated eye). This often helps distinguish between germ line and somatic cases, which has major implications for determining the risk to unaffected relatives, future siblings and offspring.
Staging
The new international classification of intra-ocular retinoblastoma has five groups – A to E of increasing severity. There is also a staging system for extraocular disease [11].
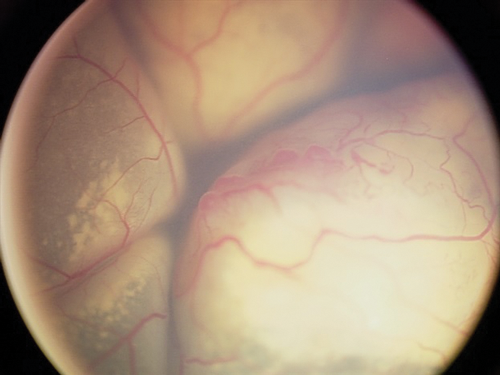
Figure 4: Lesions simulating retinoblastoma – advanced Coat’s disease.
Differential diagnosis
- Congenital cataracts – present with a white pupil (leukocoria).
- Persistent fetal vasculature (PFV), previously called persistent hyperplastic primary vitreous (PHPV), results from failure of regression of fetal vessels in the vitreous.
- Retinal dysplasia seen in Norrie’s disease, Patau’s syndrome, Edward’s syndrome, Walker Warburg and other neuronal migration disorders.
- Early onset Coat’s disease – a vascular retinopathy resulting in exudation of lipid under the retina (Figure 4).
- Congenital infections like Toxocara affecting the eye.
- Congenital glaucoma presenting with a cloudy, red and watery eye.
These can easily be distinguished from retinoblastoma by ophthalmologists with experience in retinoblastoma management.
References
1. Abramson DH, Servodidio CA. Retinoblastoma in the first year of life. Ophthalmic Paediatr Genet 1992;13(4):191-203.
2. Leal-Leal CA, Rivera-Luna R, Flores-Rojo M, et al. Survival in extra-orbital metastatic retinoblastoma:treatment results. Clin Transl Oncol 2006;8(1):39-44.
3. Rodriguez-Galindo C, Wilson MW, Chantada G, et al. Retinoblastoma: one world, one vision. Pediatrics 2008;122(3):e763-70.
4. Harbour JW. Eye cancer: unique insights into oncogenesis: the Cogan Lecture. Invest Ophthalmol Vis Sci 2006;47(5):1736-45.
5. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 1971;68:820-3.
6. Corson TW, Gallie BL. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes Chromosomes Cancer 2007;46(7):617-34.
7. Batra R, Rowe F, Rowlands A, Noonan C. Unilateral leucocoria in clinically normal eyes. Br J Ophthalmol 2009;93(4):556-7.
8. Leander C, Fu LC, Pena A, et al. Impact of an education program on late diagnosis of retinoblastoma in Honduras. Pediatr Blood Cancer 2007;49:817-9.
9. Shields CL, Honavar S, Shields JA, et al. Vitrectomy in eyes with unsuspected retinoblastoma. Ophthalmology 2000;107(12):2250-5. 16. Shields JA, Shields CL, Parsons HM. Differential diagnosis of retinoblastoma. Retina 1991;11(2):232-43.
10. Kaufman LM, Mafee MF, Song CD. Retinoblastoma and simulating lesions. Role of CT, MR imaging and use of Gd-DTPA contrast enhancement. Radiol Clin North Am 1998;36(6):1101-17.
11. Linn Murphree A. Intraocular retinoblastoma: the case for a new group classification. Ophthalmol Clin North Am 2005;18(1):41-53.
Declaration of competing interests: None declared.
See here for Part 2 of this topic.
COMMENTS ARE WELCOME








