Based on biomicroscopic and histopathologic features, corneal dystrophies can be broadly classified into epithelial, Bowman’s layer / anterior stromal, stromal, and endothelial dystrophies.
Corneal dystrophies are a group of inherited, bilateral, gradually progressing, non- inflammatory conditions caused by accumulation of extraneous material in the cornea. Changes usually begin in one of the layers of cornea and gradually spread to other layers. Patients can be asymptomatic in the early stages and later on present with symptoms of recurrent corneal erosion or visual symptoms depending on the corneal layer involved and extent of spread.
Corneal dystrophies do turn up in Objective Structured Clinical Examinations (OSCE) exams. Some tips to identify them include realising a graft in the fellow eye, recurrence of disease on the graft and bilaterality (although more common, not a rule). The questions following this can either be about the likely dystrophy, inheritance pattern, clinical features and management, or questions on the types of corneal graft procedures, indications, complications, features of rejection and management.
In this article I will briefly describe the classification of corneal dystrophies, salient features of common dystrophies and a brief outline on management.
Based on biomicroscopic and histopathologic features, corneal dystrophies can be broadly classified into epithelial, Bowman’s layer / anterior stromal, stromal, and endothelial dystrophies. The international committee for classification of corneal dystrophies (IC3D) classifies corneal dystrophies incorporating new genetic, clinical and pathological information into the traditional system of classification [1].
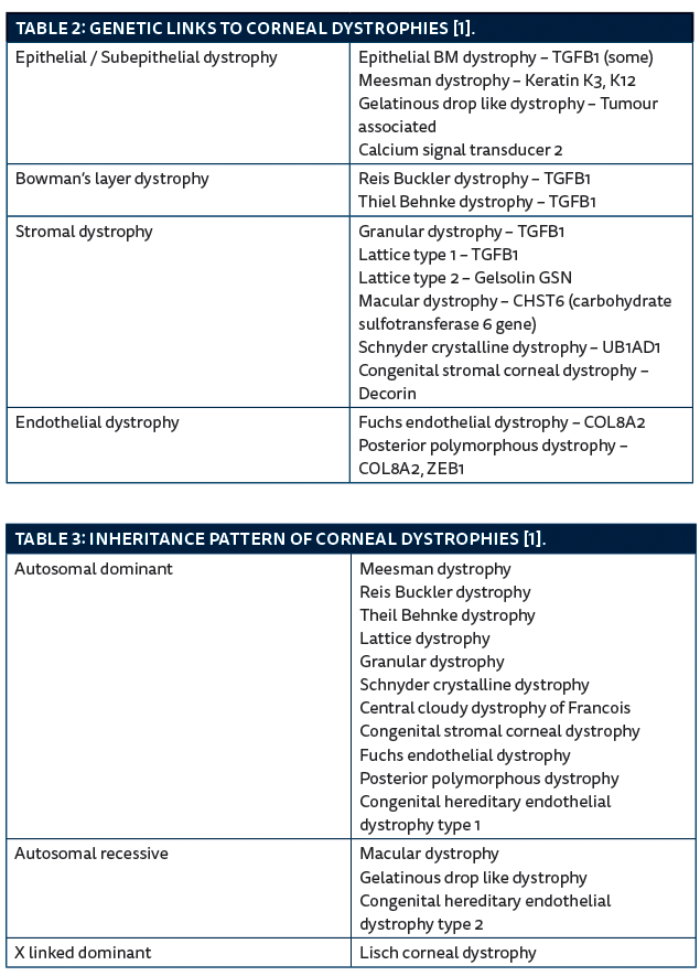
The next section describes the salient features of these corneal dystrophies followed by tables showing genetic links, inheritance patterns and some take home tips for trainees.
Epithelial dystrophies
Epithelial basement membrane dystrophy / Map dot fingerprint dystrophy / Cogan’s microcystic dystrophy:
This is characterised by presence of irregular islands of thickened grey epithelium (maps), irregular round or comma shaped putty grey patches (dots), and fine refractile parallel curvilinear lines (fingerprints) in the epithelium [1]. There is a risk of recurrent corneal erosions. Onset is in adulthood, there is no inheritance pattern recognised although in some patients a defect in the TGFB1 gene has been noted.
Meesman dystrophy:
This rare dystrophy usually manifests by the first year of life, but patients are asymptomatic until middle age. There are discrete, tiny, epithelial vesicles all over cornea, extending to the limbus but more concentrated in the palpebral fissure. It is inherited in an autosomal dominant pattern. The gene defect has been identified in the keratin K and K12 genes.
Lisch corneal dystrophy:
It is characterised by localised grey opacities in the epithelium in different patterns which can be whorl-like, radial, band shaped, feathery or flame shaped. Patients are usually asymptomatic unless the deposits are in the visual axis. It is inherited in an X-linked dominant manner.
Gelatinous drop-like dystrophy:
This is characterised by subepithelial nodular deposits which later on involve the stroma and gradually increase in size giving a mulberry-like appearance. It has been noted subepithelial amyloidosis deposits can be found in the basal epithelial layers. It is inherited in an autosomal recessive pattern with a defect in the tumour associated calcium signal transducer 2 gene. Age of onset is in the first to second decade. Superficial corneal vascularisation is frequently seen.
Bowman layer / anterior stromal dystrophy
Reis Buckler dystrophy:
This is characterised by presence of grey white, fine, round, subepithelial opacities, these are most dense centrally. It is inherited in an autosomal dominant pattern; the defect is on the TGFB1 gene. Onset is usually during childhood, the deposit increases in density with age. Vision is affected because of scarring at Bowman’s layer level.
Thiel-Behnke (honey comb) dystrophy:
With onset at the end of first decade, patients with this dystrophy usually present with symptoms of recurrent corneal erosion. Visual acuity is usually good. It is characterised by subepithelial opacities in a honeycomb pattern, mainly involving the central cornea. Inheritance pattern is autosomal dominant with the genetic defect on the TGFB1 gene.
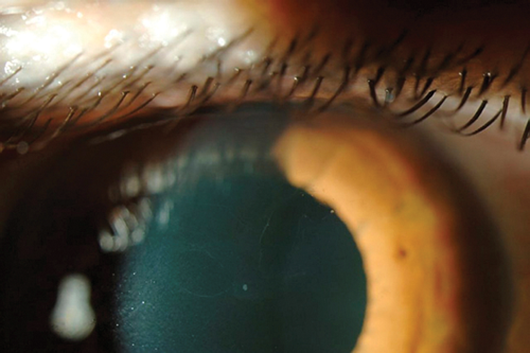
Figure 1: Right Map dot dystrophy, note the typical islands.
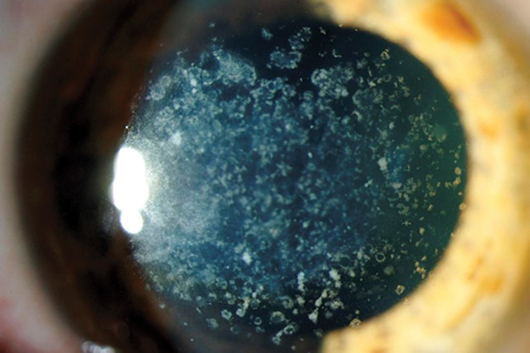
Figure 2: Granular dystrophy.
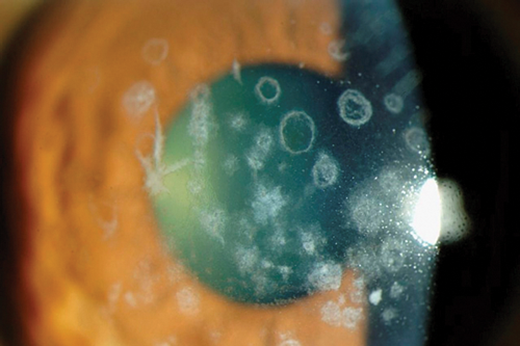
Figure 3: Avellino dystrophy.
Stromal dystrophies
Lattice corneal dystrophy:
Type 1: This is characterised by presence of thin branching refractile lines within the stroma, sparing the limbus and gradually leading to generalised stromal haze. Histology shows amyloid deposits in the stroma which stains with Congo red. The lines start appearing at the end of the first decade. They start centrally and superficially and then spread centrifugally. It then involves the deeper layers, sparing the peripheral 1mm, Descemet’s membrane and endothelium. It is inherited in an autosomal dominant pattern; the defect is linked to the TGFB1 gene.
Type 2 or Meretoja syndrome is characterised by sparse lines. There are features of systemic amyloidosis with bilateral cranial and peripheral neuropathy. The corneal lines spread centripetally from the limbus, the central cornea is spared.
Type 3 is characterised by late onset at the age of 70-90 years with thick rope-like bands.
Granular dystrophy:
It begins in childhood. It is characterised by small white sharply demarcated deposits resembling crumbs. Initially the stroma between the opacities is clear, but the deposits gradually increase in size and number and confluence leading to diffuse haze. The limbus is typically spared. Histology shows hyaline deposits staining with Masson’s trichrome. This is another corneal dystrophy linked to the TGFB1 gene. It is inherited in an autosomal dominant pattern.
Avellino dystrophy:
This is a combined Granular and Lattice dystrophy. Histology shows both hyaline and amyloid deposits in the stroma. It usually manifests in the second decade, with refractile lines and granular deposits, recurrent erosions are rare.
Macular dystrophy:
This dystrophy represents an in-born error of keratin sulphate metabolism. Onset is at the end of the first decade. Patients usually present with reduced vision. It is inherited in an autosomal recessive pattern. The genetic defect is on the carbohydrate sulfotransferase 6 gene (CHST6). Clinically there are poorly delineated deposits in the central corneal stroma with surrounding anterior stromal haze. The deposits gradually increase in size involving the full thickness of stroma up to the limbus. Histology shows deposits of glycosaminoglycan in the stroma which stains with alcian blue.
Schnyder crystalline corneal dystrophy: This dystrophy represents a defect in corneal lipid metabolism. Fifty percent of patients have raised serum cholesterol levels. Histology shows deposits of phospholipids and cholesterol. Onset is in the second decade. It is characterised by subepithelial crystalline deposits, eventually there is diffuse corneal haze and prominent corneal arcus by the third decade. The genetic defect is on the UB1AD1 gene with autosomal dominant inheritance.
Central cloudy dystrophy of Francois: This has similar appearance to crocodile shagreen with polygonal cloudy grey opacities in posterior stroma giving a ‘leather-like appearance’. The difference from crocodile shagreen is that Francois dystrophy is located in a central and posterior location. It is inherited in an autosomal dominant pattern. Patients are usually asymptomatic.
Congenital stromal corneal dystrophy (Whitschel dystrophy)
This dystrophy is very rare. It is present at birth. It consists of numerous opaque flaky or feathery areas of clouding in the stroma that multiply with age. There is some association with primary open glaucoma. The mutation is on the DCN gene on chromosome 12q22 that encodes the protein decorin. It is inherited in an autosomal dominant manner.
Endothelial dystrophy
Fuchs endothelial dystrophy:
This dystrophy usually manifests after the age of 50, it is more common in females. The initial findings are corneal guttata giving a ‘beaten metal appearance’, later there may be endothelial failure and stromal oedema. Patients usually present with blurred vision worse in the mornings. Persistent epithelial oedema leads to micro cysts and bullae which rupture giving pain and discomfort. This dystrophy can be associated with open angle glaucoma.
Posterior polymorphous corneal dystrophy:
This is characterised by vesicles arranged in a linear or grouped pattern, the appearance is of broad bands with irregular, scalloped edges. This can be associated with iris abnormalities and glaucoma. Onset during early childhood, the genetic defect is linked to COL8A2 (type 1) and ZEB1 (type 3) with autosomal dominant inheritance.
Congenital hereditary endothelial dystrophy:
Patients with this dystrophy usually present with bilateral corneal oedema. Autosomal recessive form (type 2) manifests at birth and is non progressive, whereas the autosomal dominant form (type 1) presents during childhood and is slowly progressive.
Management of corneal dystrophies
The management is generally based on symptoms. If there are signs of recurrent corneal erosion, in the acute management topical antibiotics, padding the eye or bandage contact lenses and lubricants are used. Long-term lubricants or bandage contact lenses are used for extended periods of time in frequent recurrences. If excessive episodes of erosions or a reduction in visual acuity, phototherapeutic (excimer laser) keratectomy can be effective, it removes the superficial opacities, smoothes the surface and allows the epithelium to re-adhere more tightly.
If there is worsening vision or deeper opacification, patients will benefit from lamellar or full thickness corneal transplantation. For dystrophies involving the epithelium or superficial stroma, superficial anterior lamellar keratoplasty (SALK) is found to be beneficial, while deep anterior lamellar keratoplasty (DALK) is advised for deeper stromal involvement. Most of the endothelial dystrophies can be treated with Descemet’s stripping endothelial keratoplasty (DSEK) or Descemet’s membrane endothelial keratoplasty (DMEK) but some patients might need a full thickness penetrating keratoplasty if they have significant stromal or epithelial scarring. Most of the dystrophies are found to recur in the graft after a few years.
References
1. Weiss JS, Moller H, Lisch W, et al. The IC3D classification of corneal dystrophies. Cornea 2008;27(10:suppl2):S1-S42.
2. American Academy of Ophthalmology. External diseases and cornea. 2013-2014.
TAKE HOME MESSAGE
-
The most common gene associated with corneal dystrophies is TGFB1.
-
All stromal corneal dystrophies are inherited in an autosomal dominant pattern except for macular dystrophy which is autosomal recessive.
-
Most of the corneal dystrophies manifest clinically during childhood or in the first decade, except Map dot fingerprint dystrophy and Fuchs dystrophy, which start manifesting later in life.
-
Most of the stromal dystrophies spare the limbus, except for macular dystrophy which involves the limbus.
-
Schnyder crystalline dystrophy presents with prominent arcus in young patients and can be associated with hyperlipidaemia.
-
Central cloudy dystrophy of Francois clinically resembles crocodile shagreen.
-
The common stromal dystrophies and the type of deposit within it can be remembered using the mnemonic below:
Marilyn – Macular dystrophy
Monroe – Mucopolysaccharide
Always – Alcian blue
Gets – Granular dystrophy
Her – Hyaline
Man – Masson’s trichrome
in
L – Lattice dystrophy
A – Amyloid
City – Congo Red
Declaration of competing interests: None declared.
COMMENTS ARE WELCOME





