The prevalence of diabetes has continued to increase over the years. It is currently estimated that there are 382 million with diabetes worldwide in 2013, and that this figure is expected to rise to 592 million by 2035 [1]. In the United Kingdom, the prevalence of diabetes was 6.2% of the population in 2014 [2]. Diabetic retinopathy (DR) is a common complication of diabetes which is most related to the duration of diabetes, but heavily influenced by the poor control of diabetes (hyperglycaemia), associated cardiovascular risks of hypertension and hyperlipidaemia, renal disease and smoking [3-5].
Genetics is thought to play a significant role in the development of DR as well [6], although this role is not as clear as that in age-related macular degeneration. Diabetes and hyperglycaemia have significant metabolic effects on the cells of the retinal vascular, as the glucose concentration in these cells reflects that in the blood and tissue fluid. Molecular alterations occur within the retinal vascular endothelial cells and pericytes that result in increased vascular leakage (increased permeability), vascular occlusions, subsequent ischaemia and angiogenesis [3-5]. These changes manifest clinically as DR. At the same time, changes occur in the other retinal cells resulting in retinal neurodegeneration. However, this neurodegeneration is not noted clinically [7].
Normally, the retinal vascular system is designed to prevent leakage of fluid into the retinal tissue protecting it from excess fluid and potentially harmful molecules in circulation, by the inner blood-retinal barrier (BRB) formed by the tight junctions between the single layer of tightly adherent endothelial cells (ECs), their basal lamina, surrounding pericytes, astrocytes and microglia. Fluid flow from these retinal blood vessels is regulated by two main mechanisms: one involving the opening and closing of inter-endothelial ‘tight’ junctions (TJ) (paracellular pathway), and the second involving the transport vesicles that travel through the endothelial cells (transcellular pathway). A breakdown of these mechanisms of the BRB leads to increased permeability if the retinal vasculature as summarised by Klaassen et al. (2013) [7]. This breakdown results in diabetic macular oedema (DMO). The increased intraretinal fluid leads to progressive retinal dysfunction, and if left untreated will result in permanent visual loss [7]. The contributions of the choroidal vasculature to the clinical disease of DR are less well understood, but again will be largely contributed to by the choroidal EC alterations [8-11].
DMO is characterised by vascular leakage through endothelial transcellular and paracellular routes, which clinically manifests as tissue oedema and the deposition of exudates in the macula. The leakage (or DMO) may be confirmed and quantified with optical coherence tomography (OCT) and / or fundus fluorescein angiography (FFA). Diabetic macular oedema is responsible for significant visual impairment in diabetic patients [5,12-14].
There are anatomical and biochemical changes, which are interlinked, and modified by genetic factors.
Anatomic changes in DMO
Essentially, no retinal cell type is exempt from the damaging effects of hyperglycaemia in diabetes. The retinal capillary endothelial cells become leaky as described above. Retinal endothelial cell (REC) proliferation is reduced, in addition to increased death through apoptosis, which may take some time to be noticed. Similarly, there is increased pericyte loss through apoptosis, and dysfunction. The mechanism underlying pericyte apoptosis remains unclear, but has been attributed to the accumulation of stable advanced glycation end products (AGEs), abundantly found in hyperglycaemia [5,7].
There is thickening of the basement membrane (basal lamina) surrounding the REC and pericytes, whilst the endothelial cells may become thinner. An early event in the pathogenesis of diabetic vasculopathy is leukocyte adherence to retinal vascular endothelium, resulting in EC death, vascular leakage, and capillary closure [15]. After a period of time, the cell loss results in acellular capillaries, and microaneurysm formation. Occlusion of retinal capillaries and arterioles lead to retinal ischaemia and hypoxia which may progress to retinal neovascularisation, depending on severity.
The retinal astrocytes normally improve barrier properties by inducing the production of tight junction proteins. Hyperglycaemia leads to significant loss of retinal astrocytes / glial cells [7].
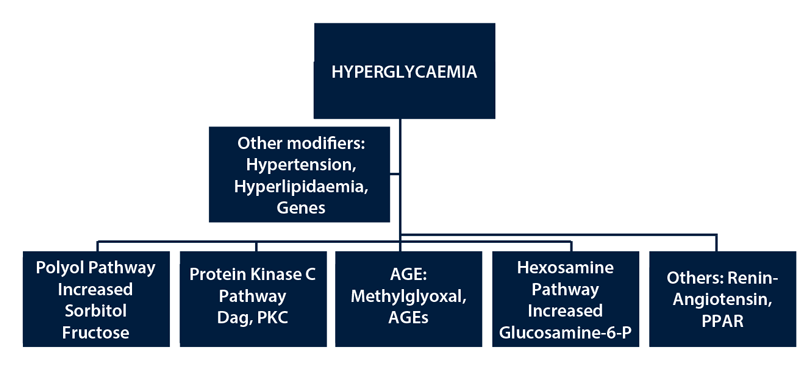
Figure 1: Pathogenesis of diabetic retinopathy.
Molecular changes in DMO
The molecular changes in DMO are due to the overproduction of reactive oxygen species (ROS) in the cellular mitochondria in the different cells, leading to oxidative stress and tissue damage through a number of major mechanisms (reviewed in Amoaku et al. 2015) [16], which are still not fully unravelled. Hyperglycaemia leads to activation of the protein kinase C (PKC) isoforms, over-activity of the hexosamine pathway, increased flux of glucose and other sugars through the polyol pathway, and increased intracellular formation of advanced glycation products (AGEs) and increased expression of the receptor for AGEs (RAGE). Other pathways include the renin-angiotensin and peroxisome proliferator-activated receptor gamma (PPAR-γ) (also known as the glitazone receptor) pathways (See Figure 1). The increased ROS may lead to inflammation by generating IL-1, IL-6, IL-8, MCP-1, iNOS, IP-10, MMPs (especially MMP9), C5-9 and TNF-alpha, as well upregulating endothelial adhesion molecules such as ICAM-1 (CD54) or E-Selectin (CD62E) [17-19], VCAM-1 (CD106) [20] and PECAM (CD31) in ECs.
The ROS directly affect the retinal neurovascular unit leading to increased breakdown of the BRB. There is significant increase in vascular endothelial growth factor (VEGF) levels from the ROS, which in turn induces increased retinal EC permeability through TJ alterations. Similarly, the increased ROS leads to increased angiopoietin 2, reduced PDGF and reduced VE-Cadherin which together result in pericyte loss. Reduced EC proliferation and increased apoptosis also result from increased ROS generation. These molecular changes and how they contribute to the increased vascular permeability in DMO are summarised in Figure 2.

Figure 2: Pathogenesis and therapeutic approaches in DMO.
It is thought that the ROS including peroxynitrite and methylglyoxal lead to increased PARP activation in the cytosol and nucleus of the retinal vascular endothelial cell, setting up a cycle that results in reduction of GADPH in the cells with the subsequent changes that manifest in clinical changes of DR (see Klaassen review) [21].
Recent evidence has confirmed that DMO is not solely due to increased VEGF levels [22]. Roh et al. (2009) [23] showed that there was increased IL-6, IL-8, VEGF, and MCP-1 significantly elevated in aqueous humour in eyes with clinically significant macular oedema (CSMO), and that the elevated IL-6, IL-8 and MCP-1 levels were noted with recurrences of CSMO after intravitreal injections of bevacizumab [23]. Similarly, Funk et al. (2010) [24] and Sohn et al. (2011) [25] have reported that IL-8, IP-10, MCP-1, and VEGF significantly higher in the aqueous humour of DMO group than in controls, and that IL-6, IP-10, MCP-1, PDGF-AA, and VEGF were significantly decreased in the eyes treated with intravitreal injections of triamcinolone. However, only VEGF reduced in the intravitreal bevacizumab treated group. Funatsu et al. (2001, 2002, 2005) [26-28] reported significant increase in vitreous ICAM-1 in eyes with DMO, increased vitreous IL-6 and VEGF in DR, increased vitreous VEGF, Angiotensin II.
It is known that retinal vascular leakage in DMO is contributed to by (VEGF) upregulation as well as non-VEGF dependent inflammatory pathways, so that chronic subclinical inflammation is important in the pathogenesis of DR [7,15,29-38]. An early event in the pathogenesis of diabetic vasculopathy is leukocyte adherence to retinal vascular endothelium, resulting in EC death, vascular leakage and capillary closure [15-16].
Treatments for DMO
Until recently, laser photocoagulation was the recommended treatment for DMO [39,40]. The exact mechanisms of action of how laser photocoagulation led to reduction in DMO are unknown. Plausible mechanisms include the destruction of high oxygen consuming photoreceptors, increased oxygenation of the retina through diffusion from the choroid though the laser scars, restoration of new retinal pigment epithelium (RPE) barrier, production of cytokines including TGF-B and pigment epithelium-derived factor (PEDF) from the stimulated RPE as discussed in the review by Bhagat et al. [5] The benefit of laser photocoagulation was only noticeable in eyes with clinically significant DMO [39,40]. The visual improvement in laser treated eyes was slow, but fairly long lasting. It is important to note, further, that photocoagulation for DMO may be associated with loss of central vision, central scotomas, and decreased colour vision. There is expansion of laser scars with time.
Subthreshold laser photocoagulation has recently been suggested as a better alternative in the treatment of DMO as the collateral damage to the retina is limited [5,41], as the subthreshold laser does not destroy the RPE the much shorter duration each application. However, the role of subthreshold laser therapy in DMO has not yet been widely accepted, and the technique and outcomes require further evaluation.
Focal laser photocoagulation still remains the standard treatment of choice in eyes with focal leakage in the macular that way from the edge of the foveal avascular zone [16].
Pharmacological treatments for DMO
Several pharmacologic agents are now available for the treatment of DMO including anti-VEGF agents and corticosteroids, and are summarised in a recent review [16]. These treatments are particularly useful in eyes with centre-involving DMO. Targeting VEGF has resulted in the use of anti-VEGFs including pegaptanib, ranibizumab, aflibercept and bevacizumab in the treatment of DMO [16]. Several clinical studies have investigated the efficacy of steroids such as triamcinolone, fluocinolone and dexamethasone in the treatment of DMO [16]. Although steroids demonstrate some anti-VEGF effects, this varies amongst the different steroids. Corticosteroids may reduce DMO mainly by targeting the non-VEGF dependent inflammatory pathways including blockage of the arachidonic acid pathway (reduction of prostaglandin synthesis, and inhibition of the release of pro-inflammatory mediators, including VEGF (see Figure 2). This effect may be through modulation of EC TJ molecules [29].
Anti-VEGF therapies
The different intravitreal VEGF inhibitors act to reduce VEGF levels in the eye and thus reverse the vascular permeability increase. Pegaptanib (Macugen, Pfizer) blocks only the VEGF-165 isoform which was originally thought to be the main pathogenic isoform. It has no effect on the other VEGF isoforms. The other available anti-VEGF agents: ranibizumab (Lucentis, Novartis), aflibercept (Eyelea, Bayer) and bevacizumab (Avastin, Roche) block all the VEGF isoforms. As other VEGF isoforms are now known to be as important as VEGF 165, it is not surprising that these other agents seem more effective in reducing DMO than pegaptanib. Ranibizumab and aflibercept have received their marketing authorisation for the treatment of DMO, whilst bevacizumab remains unlicensed for intraocular injection, and pegaptanib does not have marketing authorisation for DMO. As the effects of these anti-VEGF therapies are short lived, the DME often recurs. Furthermore, not all eyes with DMO are responsive to anti-VEGF therapies, especially if the leakage is chronic. (Chronicity of DMO is not clearly defined but is generally accepted as oedema present for six months or more).
Corticosteroids
Intravitreal triamcinolone has been used for several years to treat centre involving DMO especially when unresponsive to laser photocoagulation. However, the commonly available product, Kenalog (Squibb) remains unlicensed for intravitreal administration. The manufacturer has, in addition, issued a few ‘Dear Doctor letters’ advising against its use for that indication. Kenalog (Squibb) contains alcohol which may not be too kind to the retina. Trivaris (Allergan) is designed and licensed for intravitreal injection in the US but not in the EU. Fluocinilone (Iluvien, Alimera Sciences) and dexamethasone implant (Ozurdex, Allergan) have received marketing authorisation for the treatment of DMO, and have been approved by NICE for particular indications in DMO (in eyes that are pseudophakic). Fluocinolone (not degradable) is a slow release product thought to last up to two to three years in the vitreous, whilst Ozurdex (Allergan) (degradable) was designed for use at six monthly intervals, but is now known to require repeat injections at four to five months in most patients. These steroids show good efficacy, but are reserved as second line agents where the DMO shows suboptimal response to anti-VEGF therapies, or when anti-VEGFs are not available or suitable. It is known that as many as 20% of eyes with DMO show such suboptimal response to anti-VEGF therapies. Such poor response is more likely in eyes with chronic DMO, i.e. DMO present for an average of six months or longer. However, intravitreal steroids have increased risks of cataracts and steroid induced glaucoma which may limit their use.
Combination therapy in DMO
The available evidence suggests that each individual treatment modality in DMO does not result in a completely dry macula in most cases. Furthermore, it takes a while to achieve a dry macula in most cases, requiring frequent treatments. The ideal treatment for DMO should improve vision and improve morphological changes in the macular (for example, reduce macular oedema) for a significant duration, reduced adverse events, reduce treatment burden and costs, and be well tolerated by patients.
"Essentially, no retinal cell type is exempt from the damaging effects of hyperglycaemia in diabetes."
As discussed previously, it is now known that retinal vascular leakage in DMO is contributed to by VEGF upregulation as well as non-VEGF dependent inflammatory pathways which may be chronic and subclinical [15,29-38]. An early event in the pathogenesis of diabetic vasculopathy is leukocyte adherence to retinal vascular endothelium, which predispose to and result in EC death, vascular leakage, and capillary closure [15]. These inflammatory changes are more amenable to steroid therapies compared to anti-VEGFs.

Figure 3: Combination therapy in DMO.
As such combination of steroids and anti-VEGF therapies may provide better outcomes by blocking both the VEGF dependent and non-VEGF dependent pathways. Eyes with DMO that do not respond to monotherapy with anti-VEGF blockage should receive combinations of anti-VEGF and steroids. These may further be combined with macular laser photocoagulation, or subthreshold laser as necessary (Figure 3).
Such combinations may result in reduced treatment frequencies, better visual acuities, and less adverse events.
Abbreviations List
C = Complement
ICAM = intercellular adhesion molecule
iNOS = inducible nitric oxide synthase
IL = interleukin
IP = Interferon gamma-induced protein
MCP = monocyte chemotactic protein
MMP = matrix metalloproteinase
PECAM = Platelet endothelial cell adhesion molecule
TNF = tumour necrosis factor
VCAM = Vascular cell adhesion protein
References
1. Guariguata L, Whitting DR, Hambleton I, et al. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract 2014;103:137-49.
2. Diabetes prevalence 2014 (June 2015). Diabetes UK.
https://www.diabetes.org.uk/
About_us/What-we-say/
Statistics/Diabetes-prevalence-2014/
Last accessed August 2015.
3. Bhavsar AR. Diabetic retinopathy: the latest in current management. Retina 2006;26(6 Suppl):S71-9.
4. Bhavsar AR, Tornambe PE. 25 years of progress in the treatment of retinal diseases: where we have been, where we are now, and where we will be. Retina 2006;26(6 Suppl):S1-6.
5. Bhagat N, Grigorian RA, Tutela A, Zarbin MA. Diabetic macular edema: pathogenesis and treatment. Surv Ophthalmol 2009;54(1):1-32.
6. Kuo JZ, Wong TY, Rotter JI. Challenges in elucidating the genetics of diabetic retinopathy. JAMA Ophthalmol 2014;132:96-107.
7. Klaassen I, Van Noorden CJ, Schlingemann RO. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog Retin Eye Res 2013;34:19-48.
8. Hidayat AA, Fine BS. Diabetic choroidopathy. Light and electron microscopic observations of seven cases. Ophthalmology 1985;92(4):512-22.
9. Fryczkowski AW, Hodes BL, WalkerJ. Diabetic choroidal and iris vasculature scanning electron microscopy findings. Int Ophthalmol 1989;13(4):269-79.
10. Bischoff PM, Flower RW. Ten years experience with choroidal angiography using indocyanine green dye: a new routine examination or an epilogue? Doc Ophthalmol 1985;60(3):235-91.
11. Ishibashi T, Murata T, Kohno T, et al. Peripheral choriovitreal neovascularization in proliferative diabetic retinopathy: histopathologic and ultrastructural study. Ophthalmologica 1999;213(3):154-8.
12. Resnikoff S, Pascolini D, Etya'ale D, et al. Global data on visual impairment in the year 2002. Bull World Health Organ 2004;82(11):844-51.
13. King H, Aubert RE, Herman WH. Global burden of diabetes, 1995-2025: prevalence, numerical estimates, and projections. Diabetes Care 1998;21(9):1414-31.
14. Klein R, Marino EK, Kuller LH, et al. The Wisconsin Epidemiologic Study of Diabetic Retinopathy: XVII. The 14-year incidence and progression of diabetic retinopathy and associated risk factors in type 1 diabetes. Ophthalmology 1998;105(10):1801-15.
15. Johnson MW. Etiology and treatment of macular edema. Am J Ophthalmol 2009;147(1):11-21 e1.
16. Amoaku WM, Saker S, Stewart EA. A review of therapies for diabetic macular oedema and rationale for combination therapy. Eye (Lond) 2015 [Epub ahead of print].
17. Omi H, Okayama N, Shimizu M, et al. Participation of high glucose concentrations in neutrophil adhesion and surface expression of adhesion molecules on cultured human endothelial cells: effect of antidiabetic medicines. J Diabetes Complications 2002;16(3):201-8.
18. Morigi M, Angioletti S, Imberti B, et al. Leukocyte-endothelial interaction is augmented by high glucose concentrations and hyperglycemia in a NF-kB-dependent fashion. J Clin Invest 1998;101(9):1905-15.
19. Booth G, Stalker TJ, Lefer AM, Scalia R. Mechanisms of amelioration of glucose-induced endothelial dysfunction following inhibition of protein kinase C in vivo. Diabetes 2002;51(5):1556-64.
20. Koch AE, Halloran MM, Haskell CJ, et al. Angiogenesis mediated by soluble forms of E-selectin and vascular cell adhesion molecule-1. Nature 1995;376(6540): 517-9.
21. Brownlee M. The Pathobiology of Diabetic Complications. Diabetes 2005;54:1615-25.
22. Yoshimura T, Sonoda KH, Sugahara M, et al. Comprehensive analysis of inflammatory immune mediators in vitreoretinal diseases. PLoS One 2009;4(12):e8158.
23. Roh MI, Kim HS, Song JH, et al. Effect of intravitreal bevacizumab injection on aqueous humor cytokine levels in clinically significant macular edema. Ophthalmology 2009;116(1):80-6.
24. Funk M, Schmidinger G, Maar N, et al. Angiogenic and inflammatory markers in the intraocular fluid
of eyes with diabetic macular edema and influence of therapy with bevacizumab. Retina 2010;30(9):1412-9.
25. Sohn HJ, Han DH, Kim IT, et al. Changes in aqueous concentrations of various cytokines after intravitreal triamcinolone versus bevacizumab for diabetic macular edema. Am J Ophthalmol 2011;152(4):686-94.
26. Funatsu H, Yamashita H, Ikeda T, et al. Angiotensin II and vascular endothelial growth factor in the vitreous fluid of patients with diabetic macular edema and other retinal disorders. Am J Ophthalmol 2002;133(4):537-43.
27. Funatsu H, Yamashita H, Sakata K, et al. Vitreous levels of vascular endothelial growth factor and intercellular adhesion molecule 1 are related to diabetic macular edema. Ophthalmology 2005;112(5):806-16.
28. Funatsu H, Yamashita H, Shimizu E, et al. Relationship between vascular endothelial growth factor and interleukin-6 in diabetic retinopathy. Retina 2001;21(5):469-77.
29. Joussen AM, Poulaki V, Le ML, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J 2004;18(12):1450-2.
30. Kaji Y, Usui T, Ishida S, et al. Inhibition of diabetic leukostasis and blood-retinal barrier breakdown with a soluble form of a receptor for advanced glycation end products. Invest Ophthalmol Vis Sci 2007;48(2): 858-65.
31. Min JK, Cho YL, Choi JH, et al. Receptor activator of nuclear factor (NF)-kappaB ligand (RANKL) increases vascular permeability: impaired permeability and angiogenesis in eNOS-deficient mice. Blood 2007;109(4):1495-502.
32. Kim YM, Kim YM, Lee YM, et al. TNF-related activation-induced cytokine (TRANCE) induces angiogenesis through the activation of Src and phospholipase C (PLC) in human endothelial cells. J Biol Chem 2002;277(9):6799-805..
33. Clermont AC, Cahill M, Salti H, et al. Hepatocyte growth factor induces retinal vascular permeability via MAP-kinase and PI-3 kinase without altering retinal hemodynamics. Invest Ophthalmol Vis Sci 2006;47(6):2701-8.
34. Haynes BF, Liao HX, Patton KL. The transmembrane hyaluronate receptor (CD44): multiple functions, multiple forms. Cancer Cells 1991;3(9):347-50.
35. Trochon V, Mabilat C, Bertrand P, et al. Evidence of involvement of CD44 in endothelial cell proliferation, migration and angiogenesis in vitro. Int J Cancer 1996;66(5):664-8.
36. Schmidt AM, Hori O, Chen JX, et al. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J Clin Invest 1995;96(3):1395-403.
37. Schram MT, Chaturvedi N, Schalkwijk C, et al. Vascular risk factors and markers of endothelial function as determinants of inflammatory markers in type 1 diabetes: the EURODIAB Prospective Complications Study. Diabetes Care 2003;26(7):2165-73.
38. Adamis AP. Is diabetic retinopathy an inflammatory disease? Br J Ophthalmol 2002;86(4):363-5.
39. Photocoagulation for diabetic macular edema. Early Treatment Diabetic Retinopathy Study report number 1. Early Treatment Diabetic Retinopathy Study research group. Arch Ophthalmol 1985;103(12):1796-806.
40. Treatment techniques and clinical guidelines for photocoagulation of diabetic macular edema. Early Treatment Diabetic Retinopathy Study Report Number 2. Early Treatment Diabetic Retinopathy Study Research Group. Ophthalmology 1987;94(7):761-74.
41. Vujosevic S, Martini F, Convento E, et al. Subthreshold laser therapy for diabetic macular edema: metabolic and safety issues. Curr Med Chem 2013;20(26):3267-71.
Declaration of Competing Interests: The author has provided consultancy services to Alcon, Alimera, Allergan, Bayer, Novartis and Thrombogenics. He has received travel grants from Allergan, Bayer and Novartis, and honoraria for lectures from Alimera, Allergan, and Novartis. He has participated in clinical trials for which his institution has received funding from Allergan, Novartis and Pfizer. His institution has further received research grants from Allergan and Novartis for non-clinical studies, and CentreVue (Italy) for clinical studies.
COMMENTS ARE WELCOME



