It is fair to say that trainees and consultants who are not medical retina specialists are a bit scared of the so called retinal ‘white dot syndromes’. It is easy to understand why this is the case, as almost every uveitis specialist describes the syndromes in slightly, or even totally, different ways and often vie with each other to get their own terms accepted in published literature.
There are also arguments as to how some conditions are part of a spectrum and how some stand alone. Then there are arguments over how important each of these arguments are in turn. I will attempt to simplify this as much as possible.
The three distinct syndromes
Birdshot retinochoroidopathy
An old style shotgun loaded with birdshot pellets has a fairly wide distribution of spread that was intended to make up for the difficult to hit moving target that is a bird in flight. The dots found in this condition vary in size, as would the pellets, but are generally around a quarter to half a disc diameter oval shape with their axis pointing towards the posterior pole. The lesions are mostly concentrated at the pole itself with increasing distance from the macula resulting in an inversely proportional concentration and number of dots. They are creamy yellow and are located in the deep retina and superficial choroid and do not become pigmented, hence an earlier alternate name of ‘vitiligous retinochoroidopathy’ (see Figure 1). As the disease is posterior there is always vitritis and cystoid macular oedema is very common, while anterior chamber cells are rarer.
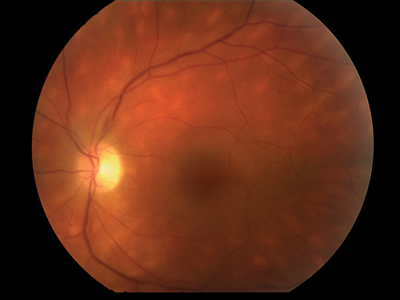
Figure 1: Birdshot retinochoridopathy.
Interestingly, it is a disease of white people almost universally with the famous association with HLA-A29 conferring a relative risk of 224 for a patient suffering from birdshot. What is fascinating, however, is that HLA-A29 itself is commonest among black South Africans and rare among whites, implying some kind of yet to be discovered trigger factor which must be at play. The condition is notoriously aggressive and usually requires equally aggressive immunosuppression. So, in, summary think of an aggressive middle-aged white person sporting a shotgun loaded with birdshot about a quarter of a disc diameter in size firing a shot at somebody’s macula.

Figure 2: Serpiginous choroiditis.
Serpiginous choroiditis
Serpiginous means ‘snake-like’ and is used in this context to describe how lesions initially form around the disc and as further lesions develop the white dots become a mass of medusa-like white snakes extending from the disc in various directions, but most commonly in an arc around the macula (see Figure 2). As it is a disease of stop-start recurrences and quiescent periods there is usually very little in the way of posterior inflammatory signs and the anterior segment is always quiet. This is indeed the main method of distinguishing it from the main differential diagnosis, toxoplasma retinochoroiditis, in which the inflammatory activity is usually very significant. As with birdshot, middle-aged white people seem to be the main group affected, although other racial groups are affected to a lesser extent.
The new creamy white extensions to the snake that form from time to time fade to a white scar, usually with pigmentation, and the aim of treatment is to suppress recurrences in order to preserve the fovea for as long as possible. Some people advocate using immunosuppression only during flare-ups in order to dampen their effect and others advocate using immunosuppression to prevent flare-ups in the first place, although the degree of immunosuppression required to do this is usually of a high order. In summary, think of another middle-aged white person with a coiled snake around their head that moves in small increments closer and closer to their terrified face.
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE)
This is a condition of younger, again mainly white people, with an average age of onset of 26 years and a slight male preponderance of 1.2:1. It is usually referred to as ‘AMPEE’ in spoken English, missing out the first ‘P’ as this renders it impossible to pronounce as a word. Unlike the other two conditions mentioned above this is a systemic condition in which a prodromal viral illness is commonly experienced and other systemic symptoms such as auditory symptoms, meningism, lymphadenopathy and bowel complaints can also occur.
‘Placoid’ means plate-like, as in a household plate, and it makes it easy then to visualise these lesions as large round yellow lesions in which the centre is slightly raised that occur at the posterior pole and are usually about half a disc to a full disc diameter in width and involve the deep retina (see Figure 3).
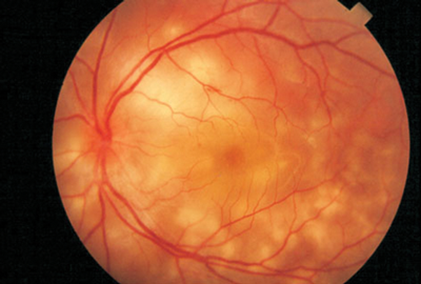
Figure 3: Acute posterior multifocal placoid pigment epitheliopathy.
The disease is self-limiting in the vast majority of cases and burns itself out in a matter of weeks with old lesions settling and new lesions appearing, not unlike a forest fire following a lightning strike at Yellowstone National Park. The lesions settle to leave a pigmented scar most of the time at the level of the retinal pigment epithelium (RPE). Much like the policy of the park firefighters at Yellowstone, the fire from spontaneous lightning strikes is usually left to burn itself out unless it shows signs of spreading to involve important park amenities, such as the fovea in this case, or to be a rare type of fire that becomes chronic and risks great destruction to the park as occurred in 1988. So the use of immunosuppression is generally discouraged unless the disease does not seem to be running its typical self-limiting course. In summary, think of a young white male dropping large yellow plates he is attempting to dry who is recovering from a viral illness and does not feel too well.
The white dot spectrum
The other white dot syndromes are perhaps most easily understood as part of a spectrum, although some do stand out and will be dealt with separately. These syndromes characteristically occur in young white myopic females predominantly and do not usually have any effective treatment or sometimes any need for treatment. This whole spectrum is sometimes referred to as ‘medical retina ornithology’, as there is much discussion about when certain birds are distinctive enough to form a species, although at the end of the day this is mostly academic, although highly interesting. After all, Bill Oddie had his own TV show.
Acute idiopathic blind spot enlargement syndrome (AIBSE)
This is the mildest condition on this spectrum and, as the name suggests, consists of a unilaterally enlarged blind spot on perimetry without a swollen disc or indeed any sign whatsoever. This condition can fluctuate over time, getting better or worse, and can rarely affect the other eye too. There is a marked relative afferent pupillary defect (RAPD) and photopsia present in the affected eye.
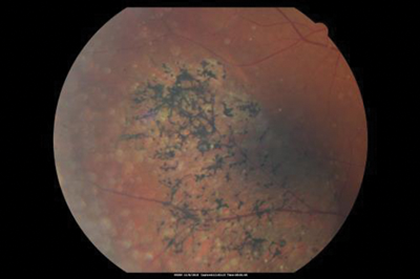
Figure 4: Acute zonal occult outer retinopathy.
Acute zonal occult outer retinopathy (AZOOR)
This condition is best thought of as a worse version of AIBSE. It is more commonly bilateral than AIBSE and the photopsia is worse, being described as being quite intrusive. The visual field defect is again an extension of the blind spot but the prognosis is more likely to be worse than better over time. Despite this, the RAPD is slightly less marked than with AIBSE. Although there is nothing to see initially, hence the word occult in the name, as time goes by there is usually some degree of RPE depigmentation and bone spicule formation in the affected area, along with retinal atrophy (see Figure 4). Indeed, retinitis pigmentosa is the main differential diagnosis in the later phase of AZOOR, as is cancer associated retinopathy and melanoma associated retinopathy in the early phase.

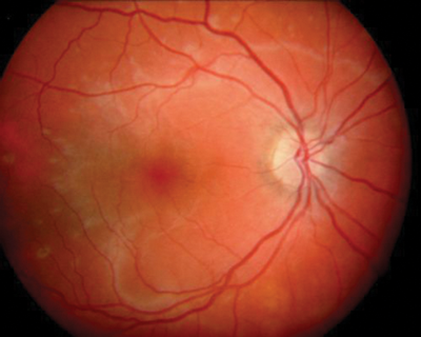
Figure 5: Acute annular outer retinopathy.
Acute annular outer retinopathy (AAOR)
This condition is similar to AZOOR in that patients present with an enlarged blind spot with photopsia but unlike AZOOR as the damage is only visible months or years after the episode has started, in the form of RPE change and retinal atrophy. Patients with AAOR present with an expanding ring of retinal inflammation centred on the disc (see Figure 5), which then expands outward leaving behind damaged retina and RPE which looks not too unlike late phase AZOOR. So AAOR can be thought of as a non-occult aggressive brush fire variant of AZOOR.
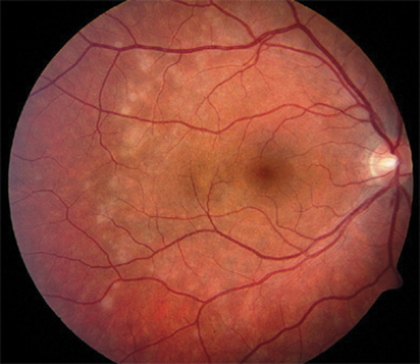
Figure 6: Multiple evanescent white dot syndrome.
Multiple evanescent white dot syndrome (MEWDS)
Evanescent is described by my Oxford English Dictionary as ‘vanishing or likely to vanish’. By far the commonest error made is to confuse ‘evanescent’ for ‘effervescent’ which implies something fizzy. These dots are not fizzy in any way, although they do disappear without a trace. It is firstly useful to consider how this condition is similar to the others in the spectrum. It is usually unilateral, is associated with an RAPD and an enlarged blind spot on perimetric testing and patients report photopsia. Unlike the others, a prodromal viral illness is common, similar perhaps to APMPEE which is outside this grouping, and of course the pathognomonic evanescent white dots occur which are not present in the other conditions (see Figure 6). These dots are smallish, around 100 microns in size, white and again, as the name suggests (now that we know what it means), they disappear. This is crucial for differentiation from other conditions that are non-evanescent, such as APMPEE, in which the lesions leave a scar. Slight posterior vitreous inflammation and periphlebitis can occur. Confusingly, these patients are said to sometimes ‘go on’ to develop AZOOR or AIBSE but in reality this phenomenon is probably further proof that these conditions all share some hidden aetiological factor and they are all different sides to a multi-sided coin.
Acute macular neuroretinopathy (AMN)
AMN is similar to AIBSE in that there is usually a unilateral scotoma, although unlike AIBSE this is located around the perifoveal area and does not represent an enlargement of the blind spot. On red free illumination of the retina wedge shaped lesions around the fovea can occasionally be seen representing a defect in the nerve fibre layer. It is possible that the oral contraceptive pill or intravenous adrenergic based chorioretinal vasoconstriction may be potential causes, although this, as with many aspects of these conditions, is the subject of debate.
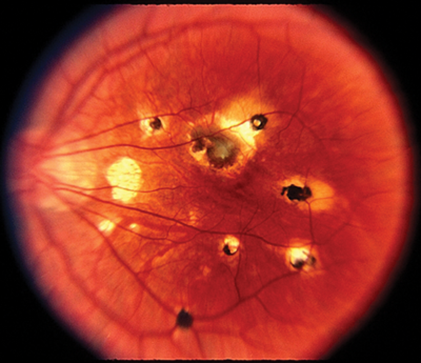
Figure 7: Multifocal choroiditis with panuveitis.
Other white dot syndromes
Multifocal choroiditis with panuveitis (MCP)
This is a bilateral inflammatory condition that, as with the spectrum described above, is most common in young myopic white women. New lesions are creamy yellow and are associated with inflammation in the form of vitreous cells and debris, cystoid macular oedema and periphlebitis. Older lesions form white punched out scars with pigmented edges and the peripapillary area is a common site for these scars, which usually measure a third to a full disc in diameter (see Figure 7). If the peripapillary lesions are fresh then a swollen disc is seen, along with impaired colour vision. As the name suggests there is potential for anterior as well as posterior segment inflammation and treating this is paramount to achieving a good outcome. In some cases it is possible to stop the immunosuppression after a period of control, while in others the chronicity of the condition is such that this is not possible. If the condition is entirely inactive and is never found to be active the main differential diagnosis is presumed ocular histoplasmosis syndrome.
Punctate inner choroidopathy (PIC)
If the lesions are few in number and are grouped around the fovea then patients are said to suffer from PIC, which is to all intents and purposes a subset of MCP. The reason that there is very little, if any, vitritis is principally due to the numbers of lesions involved rather than it being a different disease entity. With this condition, due to the perifoveolar area being so directly affected principal causes of visual morbidity stem from lesions affecting the fovea directly, which can be treated with immunosuppression if caught in the acute phase, or development of a choroidal neovascular membrane, which can be treated with anti-VEGF agents in the usual way.
Peripheral multifocal choroioretinitis
This is a subset of MCP in which there are numerous peripheral densely packed multifocal scars. Sarcoidosis is thought to be a cause of this and some authors suggest that sarcoidosis may indeed be an important aetiological factor in the conditions in the MCP spectrum.
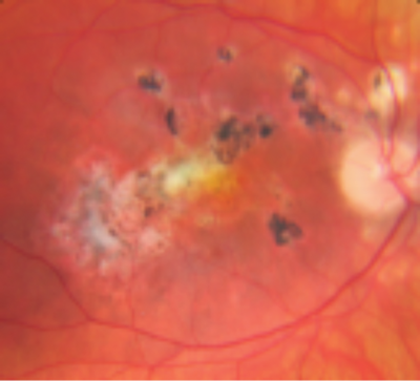
Figure 8: Progressive subretinal fibrosis and uveitis syndrome.
Progressive subretinal fibrosis and uveitis syndrome
As PIC is a small localised variant of MCP, so progressive subretinal fibrosis and uveitis syndrome is its big bad cousin from another town. MCP can progress to a stage where a subretinal gliotic sheet forms, which slowly extends across the posterior pole and if left unchecked can and will eventually involve the fovea (see Figure 8). Aggressive immunosuppression is the order of the day but even then it is very difficult to stop the lesion extending.
Retinal pigment epitheliitis (Krill’s Disease)
This condition is characterised by distorted central vision in young adults, typically after a viral infection. Examination reveals very small dark spots at the level of the outer retina, surrounded by a yellow or orange coloured halo. During the acute stage fluid may accumulate under the neurosensory retina. Over a few weeks this fluid disappears and permanent RPE scars are left behind.
Unilteral acute idiopathic maculopathy
This rare unilateral disorder occurs after a flu like illness, similar to several of the other conditions, and consists of a central retinal elevation with RPE disruption with OCT scanning revealing outer retinal thickening similar to that seen in Best’s disease, although Best’s disease is of course bilateral. Another differentiating factor is the presence of occasional vitreous cells in unilateral acute idiopathic maculopathy while again, these will be absent in Best’s disease. The majority of cases resolve quickly, although some are left with a bull’s eye lesion.
Conclusion
White dot syndromes can be a terrifying minefield of acronyms for the ophthalmic registrar of seemingly endless conditions, but once order can be established the whole visage is much less terrifying. Remembering that out of the mishmash birdshot, serpiginous and APMPEE are distinct and those which remain can be grouped into one big box, albeit untidily, is a very good start indeed. Dividing is conquering. It is interesting to postulate as to how a similar aetiology might be behind the white dot syndromes. Are they really a disease of white people or is it a huge reporting bias? This whole area is one of the last remaining ‘wild west’ reserves of ophthalmology and it is this that makes the white dot syndromes both terrifying and exciting at the same time. Some roads have been laid and various geographical features suitable for some degree of navigation have been found, and while many explorers have documented these quite well, a modern day Lewis and Clarke expedition that joins it all up has yet to take place.
Further reading
Jones N: Uveitis, Second Edition. JP Medical Publishers; London: 2012.
COMMENTS ARE WELCOME



