Part 1: Epidemiology, classification, radiology, histopathology and associations (see Part 2 here)
In this two-part series, Li Yen Goh reviews IgG4 disease and reminds us of diagnostic challenges faced.
Introduction
Immunoglobulin G4 (IgG4) disease is a recently recognised idiopathic systemic condition characterised by mass-forming lesions which histologically feature dense lymphoplasmacytic infiltration and IgG4-positive plasma cells. These lesions eventually acquire fibrosis which potentially leads to organ failure [1].
The pathogenesis of this disease is poorly understood and is thought to possibly result from an abnormal immune response against infection, allergens or tissue injury [1]. Currently, while IgG4 prominence in affected tissues are a feature of the condition, it is unclear what role IgG4 truly plays [2]. Lesions associated with IgG4-related disease in the ophthalmic regions are collectively termed ‘IgG4-related ocular disease (IgG4-ROD)’ [3].
Epidemiology and demographics of IgG4-related disease (IgG4-RD) and IgG4-ROD
Recent work from a large international cross-sectional study has established four different phenotypes of IgG4 disease: Group 1 (pancreato-hepatobiliary), Group 2 (retroperitoneal fibrosis / aorta) Group 3 (head and neck) and Group 4 (Mikulicz and systemic), each group significantly differing from other groups in terms of race, age, sex and serum IgG4 levels [4]. Analysed together, Group 3 and 4 should capture all manifestations of IgG4-ROD, although the study did find most ocular disease presented more frequently in the head and neck group; 22% versus <1% probability in the Mikulicz and systemic group. It is believed that potential differences in the triggers may results in these varied phenotypes, although the exact mechanism of this whole process remains elusive and needs further study [4]. Several different studies across populations in Japan, the USA and China have reported that the frequency of IgG4-ROD ranges from 4.0-58.8% of IgG4-RD; this wide variation could perhaps be explained by racial / geographic variation [5-8].
This cross-sectional study also suggested that East Asian individuals are more likely to be predisposed to IgG4-RD complications in the head and neck region [4]. In contrast, non-Asian cases, predominantly Caucasians (including individuals from the Indian subcontinent) have a greater predilection for pancreato-hepatobiliary disease, retroperitoneal and / or aortic disease [4]. However, due to the case submission method in the aforementioned international cross-sectional study, by individual IgG4 specialists from each country, the relative impact of environment as opposed to race was not easily discernable [4].
Involvement of the head and neck appears to be more frequent in women than men; additionally, women tend towards IgG4 disease limited to the head and neck rather than systemic disease with head and neck manifestations [4,10]. Specifically relating to IgG4-ROD, this female preponderance has been echoed; female to male ratio ranging between 2.2-1.0 to 1.0 [5,11,12]. Interestingly, in a large study of IgG4-ROD, it was found that males were significantly more likely to experience a relapse of the disease after corticosteroid therapy [5]. Individuals with head and neck disease and including IgG4-ROD tended to be younger than other forms of IgG4-RD; on average this was approximately 54 to 57 years of age compared to IgG4 pancreatitis, for example, which ranges between 58 to 69 years of age [5,11,13]. Nevertheless, IgG4-ROD has been described in children and young adults; with the youngest confirmed case being five-years-old [14-21]. In fact, the commonest presentation of any kind of IgG4-RD in children is within the orbit [22].
Some of the difficulties in identifying the true prevalence of IgG4-RD and therefore IgG4-ROD may lie in diagnostic agreement. A proportion of IgG4-ROD cases in the past are likely to have been labeled as idiopathic orbital inflammation. One study demonstrated that among cases initially classified as either chronic dacryoadenitis or orbital inflammatory pseudotumour, 39% (15 of 38) fulfilled criteria for IgG4-ROD and five were suspicious for IgG4-ROD [23]. However, with advances in investigation capabilities, cases of IgG4-ROD are now being determined more accurately and actively rather than being relegated to a diagnosis of exclusion.
Histopathology
A large international consensus by Deshpande et al. suggests diagnostic criteria, based on three main histopathological findings: 1) dense lymphoplasmacytic infiltrate; 2) storiform fibrosis (Figure 1); 3) obliterative phlebitis [24,25]. Early lesions tend towards having a more intense lymphoplasmacytic infiltrate which then culminates in a burnt-out sclerotic phase, seen in a swirling ‘storiform’ pattern [26,27]. With regards to IgG4-ROD, the demonstration of the storiform pattern of fibrosis and obliterative phlebitis is not required, as fibrosis seen in IgG4-ROD is collagenous rather than storiform and obliterative phlebitis is inconsistently seen in ocular structures [24,28].
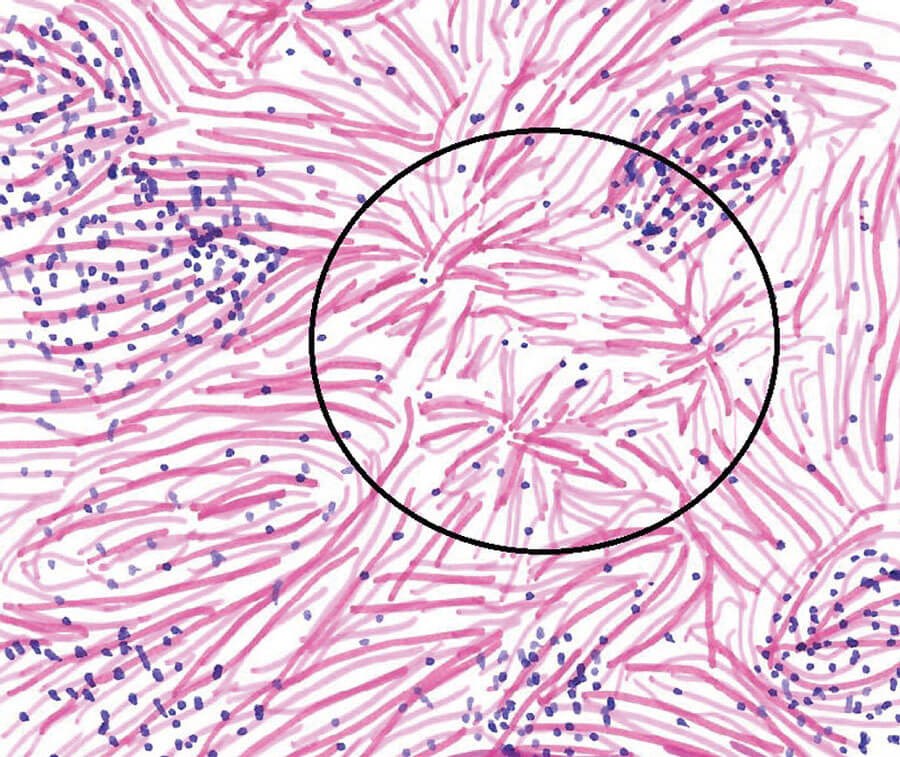
Figure 1: Illustration of storiform fibrosis (encircled) in the lacrimal gland.
Histopathological diagnosis is more difficult if needle biopsy is employed and in IgG4-ROD cases, transocular needle sampling is undertaken frequently. To optimise findings, immunohistochemistry is also undertaken routinely if IgG4 is suspected, the average number of IgG4 cells in three X40 high-powered fields (hpf) with the largest numbers should be selected [24]. Even so, needle biopsy specimens provide fewer hpfs to select from, leading to significantly lower IgG4 cells counts than surgical biopsy methods (p<0.001) [10]. Additionally, histochemical diagnostic values should be tailored towards the organ sampled but in IgG4-ROD this is not consistent; the quoted proposed cut-offs for lacrimal lesions range from as low as 10 to as high as 100 IgG4 cells per hpf [24,32]. Another well-recognised consensus by Umehara et al. from Japan was updated in 2020 and agrees broadly with the histopathological criteria set out by Deshpande et al. Additionally, this criteria also includes clinical, radiological, haematological findings in achieving a diagnosis [33]. This consensus requires clarification in terms of the size of the hpf used to count IgG4 cells. This is not uncommon, as hpf size is rarely described in papers and when it has been, the described size has varied by nearly 10-fold, between 0.0588mm2 to 0.550mm2 [34-38]. Due to these difficulties involving the hpfs, it is thought that tissue IgG4: IgG ratio may be a more powerful measure; it is preserved in needle biopsy specimens, and a ratio of >40% is thought to be a comprehensive cut-off value in any organ [40-43]. However, care needs to be taken in interpretation of these values; overall very low values of both IgG4 and IgG may result in an inflated ratio. Furthermore, in the absence of other corroborative findings of IgG4-RD, an elevated ratio could be found in multicentric Castleman’s disease, rheumatoid arthritis and other immune-mediated conditions [25-44].
Furthermore, organ specific criteria have been suggested as manifestations of IgG4 in various organs can differ. Fortunately, for ophthalmic involvement IgG4-ROD specific criteria exists and was developed by Goto et al and includes radiological, histopathological and serological findings. This criteria allows description of the disease to be classified into: definite, probable or possible IgG4-ROD [39]. View the diagnostic criteria for IgG4-related ophthalmic disease here: https://link.springer.com/article/10.1007/s10384-014-0352-2
Most recently, a joint effort between the American College of Rheumatology and the European League Against Rheumatism set out a comprehensive classification system which was based on entry criteria, exclusion criteria consisting of clinical, pathologic, serologic, and radiological findings - sensitivity of 83% and a specificity of 98.9% [45]. Importantly, even with removal of the biopsy criteria, the specificity of this classification was maintained at 94% [45]. View the 2019 American College of Rheumatology / European League Against Rheumatism classification criteria for IgG4-related disease here: https://ard.bmj.com/content/79/1/77
Serum IgG4
Elevated serum IgG4 levels alone are not diagnostic, as elevated IgG4 levels were found to be normal in as much as 40% of the population (cutoff >1.4g/L) [46,47]. This has prompted suggestions that higher optimal IgG4 cut-offs should be established but there have been mixed findings related to this, with some studies finding that even tripling the cut-off level was not discerning of IgG4 disease [47,48]. However, this test may still offer some prognostic value and aid in monitoring disease response to treatment. Associations have been demonstrated between higher IgG4 concentrations and more extensive disease, as well as increased risk of relapse [4‑6,49-54]. Interestingly, serum IgG4 tends to be higher in patients with IgG4-ROD than other IgG4-RD in other organs [5,6,10]. While IgG4 has not been established as directly involved in the pathophysiology of the disease, these total IgG4 serum levels and IgG4: IgG ratios fall with immunosuppressive treatment and corresponds to improvement in clinical features [9,55,56]. Furthermore, research has shown that lower serum IgG4 at diagnosis could be a surrogate for lower disease activity and correspondingly a lesser response to glucocorticoids [55].
Imaging
In the work-up to exclude malignancy and infection, imaging is frequently undertaken, and radiologic diagnosis based on computed tomography (CT) and magnetic resonance imaging (MRI) has been reported to have up to 80% compatibility with clinical diagnosis of IgG4-ROD [57]. Common radiologic findings on magnetic resonance imaging scans include diffuse homogenous enlargement of one / both lacrimal glands (including uniform distribution of gadolinium if used), extraocular muscle (EOM) thickening, orbital fat infiltration and trigeminal nerve enlargement. Specifically, infraorbital nerve enlargement, with bone remodeling of the infraorbital canal, especially bilaterally, is thought to be the most specific sign of IgG4 and is demonstrable on both CT and MRI scans [58-60]. Only IgG4-ROD and adnexal mucosa-associated lymphoid tissue (MALT) are known to cause enlargement of the ischaemic optic neuropathy (ION) and its canal, and both conditions have associations to one another. Almost no disease other than IgG4-ROD will lead to bilateral ION enlargement [2,58].
Lesions should be well-defined, with iso-intensity on MRI T1-weighted images and hypo-intensity on T2-weighted lesions [61]. These can easily be confused for non-specific orbital inflammation, sarcoidosis, infection, granulomatosis with polyangiitis and potentially thyroid eye disease (TED) if purely based on radiological signs, as tendon sparing is also noted in IgG4-ROD though as mentioned previously the most recent studies have noted that the lateral rectus tends to be involved more commonly in IgG4 rather than the inferior rectus in TED [62].
Associations
Lymphoma
IgG4-RD may confer a risk of developing malignancies; IgG4-ROD, dacryoadenitis, in particular may be associated non-Hodgkins lymphoma (NHL) [63]. Larger case-series suggest frequencies of between 10-14% of NHL within their populations of IgG4-ROD, notably higher than that observed in the general population [34,37,64,67]. In the general population, ocular adnexal NHL had an incidence of 0.3 per 100,000 person-years (0.3 cases arising in every 100,000 individuals in a year) [66]. The NHL most commonly associated with IgG4-ROD is MALT, this is also the most common lymphoma affecting the ocular adnexal in the general population, accounting for 35-68% of primary lymphomas [67,68]. Follicular lymphoma has also been described in association with IgG4-ROD [65]. The mechanism of lymphogenesis in IgG4-ROD and indeed IgG4-RD in general is not understood but though to be likely related to chronic antigen stimulation driving lymphoid proliferation [70].
IgE mediated disease and autoimmune disease
Associations between clinical allergy and immunoglobulin E (IgE)-mediated disease with IgG4-RD have frequently been observed though not fully understood, both are thought to have similar pathogenic mechanisms particular Th2 and IgE overproduction [71]. Studies have found the rates of allergic rhinitis to be higher in IgG4-ROD populations compared to the general population, 44% versus 34% [52,72]. Furthermore, a recent study has suggested that IgG4-ROD patients had statistically significantly higher IgE levels than patients with IgG4-RD elsewhere [5].
Conclusion
IgG4-ROD is thought to be an uncommon disease though it is recognised that its frequency may be higher than previously understood due to misdiagnosis and subsequent under-reporting, particularly in lacrimal gland and orbital involvement. Diagnosis relies on recognising histopathologic and immunohistochemical features on tissue biopsy alongside clinical assessment, serum IgG4 levels, and radiological findings.
Take home messages
-
IgG4 disease can affect almost any structure of the eye, adnexae (and body!) and therefore should be a differential in presentations of ophthalmic structure inflammation.
-
IgG4-ROD occurs most frequently among individuals aged 54-57 years and may be slightly more common in women, though men could be more prone to relapses. IgG4 disease in the head and neck are more common in East Asian individuals.
-
Diagnosis of IgG4-ROD should be based on clinical, radiological, serological, and pathological criteria, ‘possible’ and ‘probable’ diagnoses of IgG4-ROD can be allocated if the criteria are not met fully.
References
1. McNab AA, McKelvie P. IgG4-related ophthalmic disease. Part I: background and pathology. Ophthalmic Plast Reconstr Surg 2015;31(2):83-8.
2. McNab AA, McKelvie P. IgG4-Related Ophthalmic Disease. Part II: Clinical Aspects. Ophthalmic Plast Reconstr Surg 2015;31(3):167-78.
3. Stone JH, Khosroshahi A, Deshpande V, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum 2012;64(10):3061-7.
4. Wallace ZS, Zhang Y, Perugino CA, et al. Clinical phenotypes of IgG4-related disease: an analysis of two international cross-sectional cohorts. Ann Rheum Dis 2019;78(3):406-12.
5. Zhao Z, Mou D, Wang Z, et al. Clinical features and relapse risks of IgG4-related ophthalmic disease: a single-center experience in China. Arthritis Res Ther 2021;23(1):98.
6. Hamano H, Arakura N, Muraki T, et al. Prevalence and distribution of extrapancreatic lesions complicating autoimmune pancreatitis. J Gastroenterol 2006;41(12):1197-205.
7. Wallace ZS, Deshpande V, Stone JH. Ophthalmic manifestations of IgG4-related disease: single-center experience and literature review. Semin Arthritis Rheum 2014;43(6):806-17.
8. Takuma K, Kamisawa T, Anjiki H, et al. Metachronous extrapancreatic lesions in autoimmune pancreatitis. Intern Med 2010;49(6):529-33.
9. Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001;344(10):732-8.
10. Zen Y, Nakanuma Y. IgG4-related disease: a cross-sectional study of 114 cases. Am J Surg Pathol 2010;34(12):1812-9.
11. Andrew N, Kearney D, Selva D. IgG4-related orbital disease: a meta-analysis and review. Acta Ophthalmol 2013;91(8):694-700.
12. Yu WK, Tsai CC, Kao SC, Liu CJ. Immunoglobulin G4-related ophthalmic disease. Taiwan J Ophthalmol 2018;8(1):9-14.
13. Wu A, Andrew NH, McNab AA, Selva D. IgG4-Related Ophthalmic Disease: Pooling of Published Cases and Literature Review. Curr Allergy Asthma Rep 2015;15(6):27.
14. Das D, Deka P, Verma G, et al. IgG4-related intraocular inflammation masquerading as ciliary body melanoma in a young girl. Indian J Ophthalmol 2016;64(8):601-3.
15. Griepentrog GJ, Vickers RW, Karesh JW, et al. A clinicopathologic case study of two patients with pediatric orbital IgG4-related disease. Orbit 2013;32(6):389‑91.
16. Notz G, Intili A, Bilyk JR. IgG4-related dacryoadenitis in a 13-year-old girl. Ophthalmic Plast Reconstr Surg 2014;30(6):e161-3.
17. Sane M, Chelnis J, Kozielski R, Fasiuddin A. Immunoglobulin G4-related sclerosing disease with orbital inflammation in a 12-year-old girl. J AAPOS 2013;17(5):548-50.
18. Kalapesi FB, Garrott HM, Moldovan C, et al. IgG4 orbital inflammation in a 5-year-old child presenting as an orbital mass. Orbit 2013;32(2):137-40.
19. Tille L, Schnabel A, Laass MW, et al. Orbital inflammation and colitis in pediatric IgG4-related disease: A case report and review of the literature. Eur J Rheumatol 2019;7(Suupl 1):1-7.
20. Smerla RG, Rontogianni D, Fragoulis GE. Ocular manifestations of IgG4-related disease in children. More common than anticipated? Review of the literature and case report. Clin Rheumatol 2018;37(6):1721-7.
21. Kaya Akca Ü, Atalay E, Kasap Cüceoğlu M, et al. IgG4-related disease in pediatric patients: a single-center experience. Rheumatol Int 2021;42(7):1177‑85.
22. Karim F, Loeffen J, Bramer W, et al. IgG4-related disease: a systematic review of this unrecognized disease in pediatrics. Pediatr Rheumatol Online J 2016;14(1):18.
23. Ferry JA, Klepeis V, Sohani AR, et al. IgG4-related Orbital Disease and Its Mimics in a Western Population. Am J Surg Pathol 2015;39(12):1688-700.
24. Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol 2012;25(9):1181-92.
25. Strehl JD, Hartmann A, Agaimy A. Numerous IgG4-positive plasma cells are ubiquitous in diverse localised non-specific chronic inflammatory conditions and need to be distinguished from IgG4-related systemic disorders. J Clin Pathol 2011;64(3):237-43.
26. Cheuk W, Yuen HK, Chan JK. Chronic sclerosing dacryoadenitis: part of the spectrum of IgG4-related Sclerosing disease? Am J Surg Pathol 2007;31(4):643‑5.
27. Divatia M, Kim SA, Ro JY. IgG4-related sclerosing disease, an emerging entity: a review of a multi-system disease. Yonsei Med J 2012;53(1):15-34.
28. Smyrk TC. Pathological features of IgG4-related sclerosing disease. Curr Opin Rheumatol 2011;23(1):74-9.
29. Ferry JA, Deshpande V. IgG4-related disease in the head and neck. Semin Diagn Pathol 2012;29(4):235-44.
30. Mudhar HS, Bhatt R, Sandramouli S. Xanthogranulomatous variant of immunoglobulin G4 sclerosing disease presenting as ptosis, proptosis and eyelid skin plaques. Int Ophthalmol 2011;31(3):245-8.
31. Singh K, Rajan KD, Eberhart C. Orbital necrobiotic xanthogranuloma associated with systemic IgG4 disease. Ocul Immunol Inflamm 2010;18(5):373-8.
32. Umehara H, Okazaki K, Masaki Y, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol 2012;22(1):21-30.
33. Umehara H, Okazaki K, Kawa S, et al. The 2020 revised comprehensive diagnostic (RCD) criteria for IgG4-RD. Mod Rheumatol 2021;31(3):529-33.
34. Kubota T, Moritani S, Katayama M, Terasaki H. Ocular adnexal IgG4-related lymphoplasmacytic infiltrative disorder. Arch Ophthalmol 2010;128(5):577-84.
35. Wallace ZS, Khosroshahi A, Jakobiec FA, et al. IgG4-related systemic disease as a cause of “idiopathic” orbital inflammation, including orbital myositis, and trigeminal nerve involvement. Surv Ophthalmol 2012;57(1):26-33.
36. Plaza JA, Garrity JA, Dogan A, et al. Orbital inflammation with IgG4-positive plasma cells: manifestation of IgG4 systemic disease. Arch Ophthalmol 2011;129(4):421-8.
37. Go H, Kim JE, Kim YA, et al. Ocular adnexal IgG4-related disease: comparative analysis with mucosa-associated lymphoid tissue lymphoma and other chronic inflammatory conditions. Histopathology 2012;60(2):296-312.
38. Kubota T, Moritani S, Yoshino T, et al. Ocular adnexal marginal zone B cell lymphoma infiltrated by IgG4-positive plasma cells. J Clin Pathol 2010;63(12):1059-65.
39. Goto H, Takahira M, Azumi A, Disease. JSGfI-RO. Diagnostic criteria for IgG4-related ophthalmic disease. Jpn J Ophthalmol 2015;59(1):1-7.
40. Liu Y, Yang F, Chi X, et al. Needle biopsy compared with surgical biopsy: pitfalls of small biopsy in histologial diagnosis of IgG4-related disease. Arthritis Res Ther 2021;23(1):54.
41. Cheuk W, Chan JK. IgG4-related sclerosing disease: a critical appraisal of an evolving clinicopathologic entity. Adv Anat Pathol 2010;17(5):303-32.
42. Cheuk W, Chan JK. Lymphadenopathy of IgG4-related disease: an underdiagnosed and overdiagnosed entity. Semin Diagn Pathol 2012;29(4):226-34.
43. Sato Y, Kojima M, Takata K, et al. Systemic IgG4-related lymphadenopathy: a clinical and pathologic comparison to multicentric Castleman’s disease. Mod Pathol 2009;22(4):589-99.
44. Sato Y, Kojima M, Takata K, et al. Multicentric Castleman’s disease with abundant IgG4-positive cells: a clinical and pathological analysis of six cases. J Clin Pathol 2010;63(12):1084-9.
45. Wallace ZS, Naden RP, Chari S, et al. The 2019 American College of Rheumatology/European League Against Rheumatism classification criteria for IgG4-related disease. Ann Rheum Dis 2020;79(1):77-87.
46. Sah RP, Chari ST. Serologic issues in IgG4-related systemic disease and autoimmune pancreatitis. Curr Opin Rheumatol 2011;23(1):108-13.
47. Carruthers MN, Khosroshahi A, Augustin T, et al. The diagnostic utility of serum IgG4 concentrations in IgG4-related disease. Ann Rheum Dis 2015;74(1):14-8.
48. Yu KH, Chan TM, Tsai PH, et al. Diagnostic Performance of Serum IgG4 Levels in Patients With IgG4-Related Disease. Medicine (Baltimore) 2015;94(41):e1707.
49. Igarashi H, Ito T, Oono T, et al. Relationship between pancreatic and / or extrapancreatic lesions and serum IgG and IgG4 levels in IgG4-related diseases. J Dig Dis 2012;13(5):274-9.
50. Kamisawa T, Okamoto A, Funata N. Clinicopathological features of autoimmune pancreatitis in relation to elevation of serum IgG4. Pancreas 2005;31(1):28-31.
51. Masaki Y, Kurose N, Yamamoto M, et al. Cutoff Values of Serum IgG4 and Histopathological IgG4+ Plasma Cells for Diagnosis of Patients with IgG4-Related Disease. Int J Rheumatol 2012;2012:580814.
52. Matsui S, Taki H, Shinoda K, et al. Respiratory involvement in IgG4-related Mikulicz’s disease. Mod Rheumatol 2012;22(1):31-9.
53. Tsang KFP, Oppong WK, Leeds SJ, et al. Does IgG4 level at the time of diagnosis correlate with disease outcome in IgG4-Related disease? Pancreatology 2019;19(1):177-81.
54. Culver EL, Sadler R, Simpson D, et al. Elevated Serum IgG4 Levels in Diagnosis, Treatment Response, Organ Involvement, and Relapse in a Prospective IgG4-Related Disease UK Cohort. Am J Gastroenterol 2016;111(5):733-43.
55. Yu WK, Kao SC, Yang CF, et al. Ocular adnexal IgG4-related disease: clinical features, outcome, and factors associated with response to systemic steroids. Jpn J Ophthalmol 2015;59(1):8-13.
56. Tabata T, Kamisawa T, Takuma K, et al. Serial changes of elevated serum IgG4 levels in IgG4-related systemic disease. Intern Med 2011;50(2):69-75.
57. Lee MJ, Hamilton BE, Pettersson D, et al. Radiologic imaging shows variable accuracy in diagnosing orbital inflammatory disease and assessing its activity. Sci Rep 2020;10(1):21875.
58. Ohshima K, Sogabe Y, Sato Y. The usefulness of infraorbital nerve enlargement on MRI imaging in clinical diagnosis of IgG4-related orbital disease. Jpn J Ophthalmol 2012;56(4):380-2.
59. Soussan JB, Deschamps R, Sadik JC, et al. Infraorbital nerve involvement on magnetic resonance imaging in European patients with IgG4-related ophthalmic disease: a specific sign. Eur Radiol 2017;27(4):1335-43.
60. Jayaprakasam A, O’Donovan D, Rene C. Infraorbital nerve enlargement due to IgG4-related disease. Eye (Lond) 2014;28(5):628-9.
61. Song YS, Choung HK, Park SW, et al. Ocular adnexal IgG4-related disease: CT and MRI findings. Br J Ophthalmol 2013;97(4):412-8.
62. Tiegs-Heiden CA, Eckel LJ, Hunt CH, et al. Immunoglobulin G4-related disease of the orbit: imaging features in 27 patients. AJNR Am J Neuroradiol 2014;35(7):1393-7.
63. Yamamoto M, Takahashi H, Tabeya T, et al. Risk of malignancies in IgG4-related disease. Mod Rheumatol 2012;22(3):414-8.
64. Sato Y, Ohshima K, Ichimura K, et al. Ocular adnexal IgG4-related disease has uniform clinicopathology. Pathol Int 2008;58(8):465-70.
65. Cheuk W, Yuen HK, Chan AC, et al. Ocular adnexal lymphoma associated with IgG4+ chronic sclerosing dacryoadenitis: a previously undescribed complication of IgG4-related sclerosing disease. Am J Surg Pathol 2008;32(8):1159-67.
66. Moslehi R, Devesa SS, Schairer C, Fraumeni JF. Rapidly increasing incidence of ocular non-hodgkin lymphoma. J Natl Cancer Inst 2006;98(13):936-9.
67. McKelvie PA. Ocular adnexal lymphomas: a review. Adv Anat Pathol 2010;17(4):251-61.
68. Hsu CR, Chen YY, Yao M, et al. Orbital and ocular adnexal lymphoma: a review of epidemiology and prognostic factors in Taiwan. Eye (Lond) 2021;35(7):1946-53.
69. Sato Y, Notohara K, Kojima M, et al. IgG4-related disease: historical overview and pathology of hematological disorders. Pathol Int 2010;60(4):247-58.
70. Kanda G, Ryu T, Shirai T, et al. Peripheral T-cell lymphoma that developed during the follow-up of IgG4-related disease. Intern Med 2011;50(2):155-60.
71. Michailidou D, Schwartz DM, Mustelin T, Hughes GC. Allergic Aspects of IgG4-Related Disease: Implications for Pathogenesis and Therapy. Front Immunol 2021;12:693192.
72. Yonekura S, Okamoto Y, Horiguchi S, et al. Effects of aging on the natural history of seasonal allergic rhinitis in middle-aged subjects in South Chiba, Japan. Int Arch Allergy Immunol 2012;157(1):73-80.
COMMENTS ARE WELCOME





