History
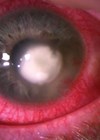
A 21-month-old boy presented to his local ophthalmology department with a left proptotic eye from a growing cystic lesion known to be present from birth. Notes taken on presentation were:
- Known left microphthalmia with chorio-retinal coloboma, contralateral eye was normal.
- General health, development and physical examination were otherwise normal.
- The cyst was removed and contained yellow, translucent fluid.
- The cyst wall was sent to the ophthalmic pathology department for assessment.
Questions
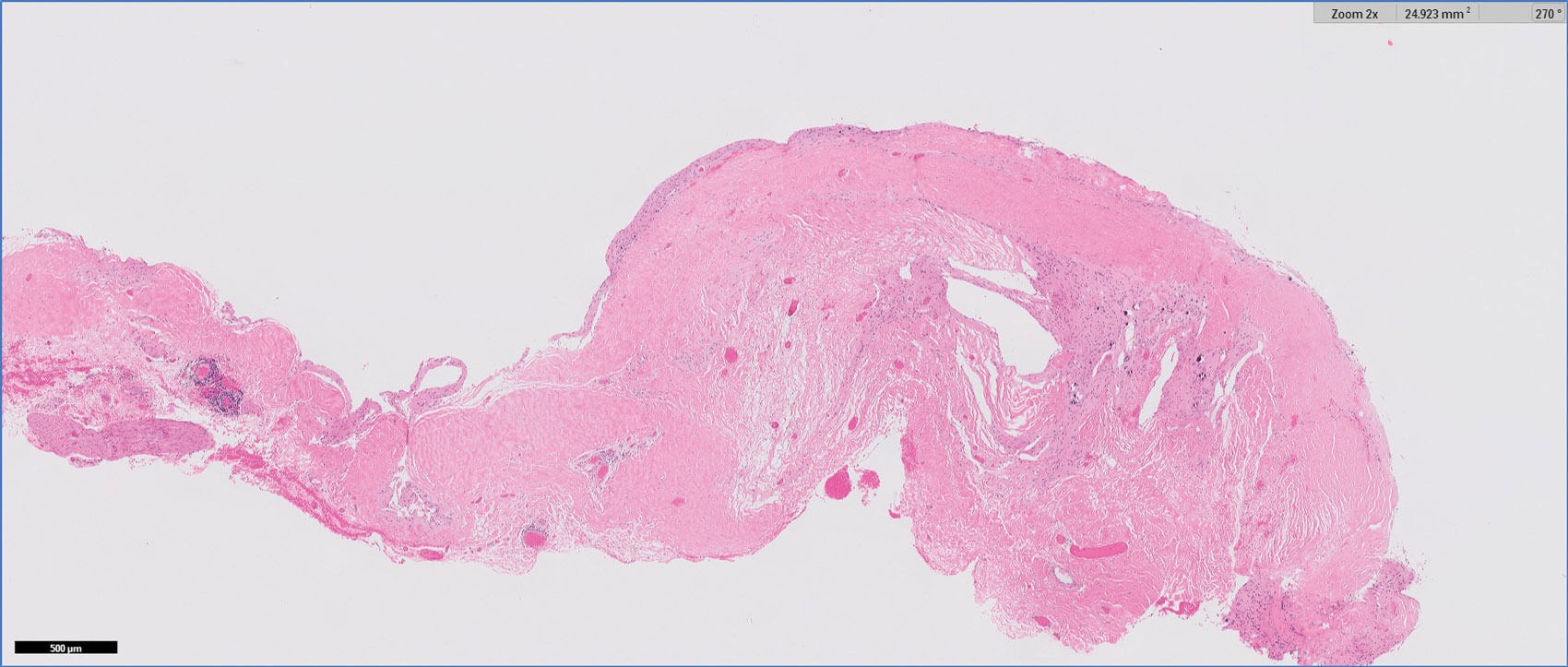
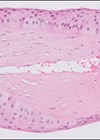
Figure 1: H&E 2x.
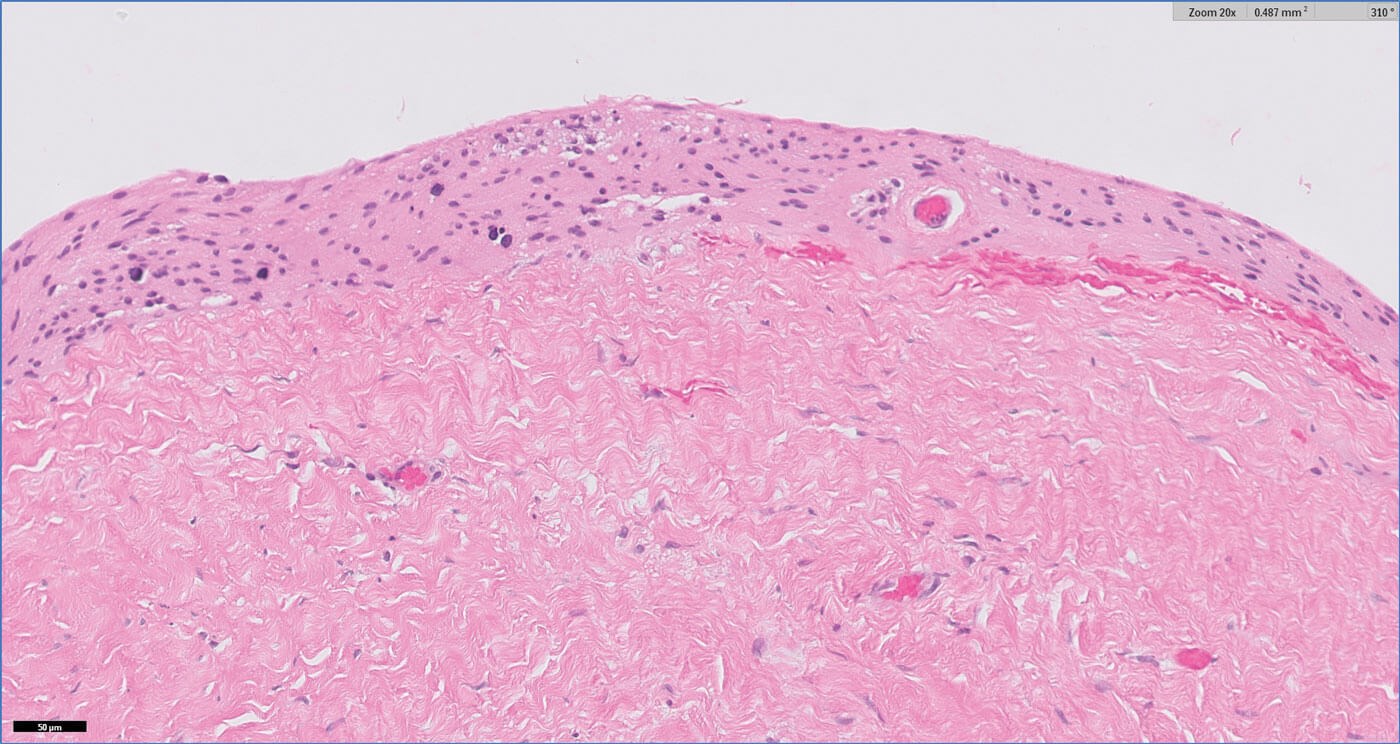
Figure 2: H&E 20x.
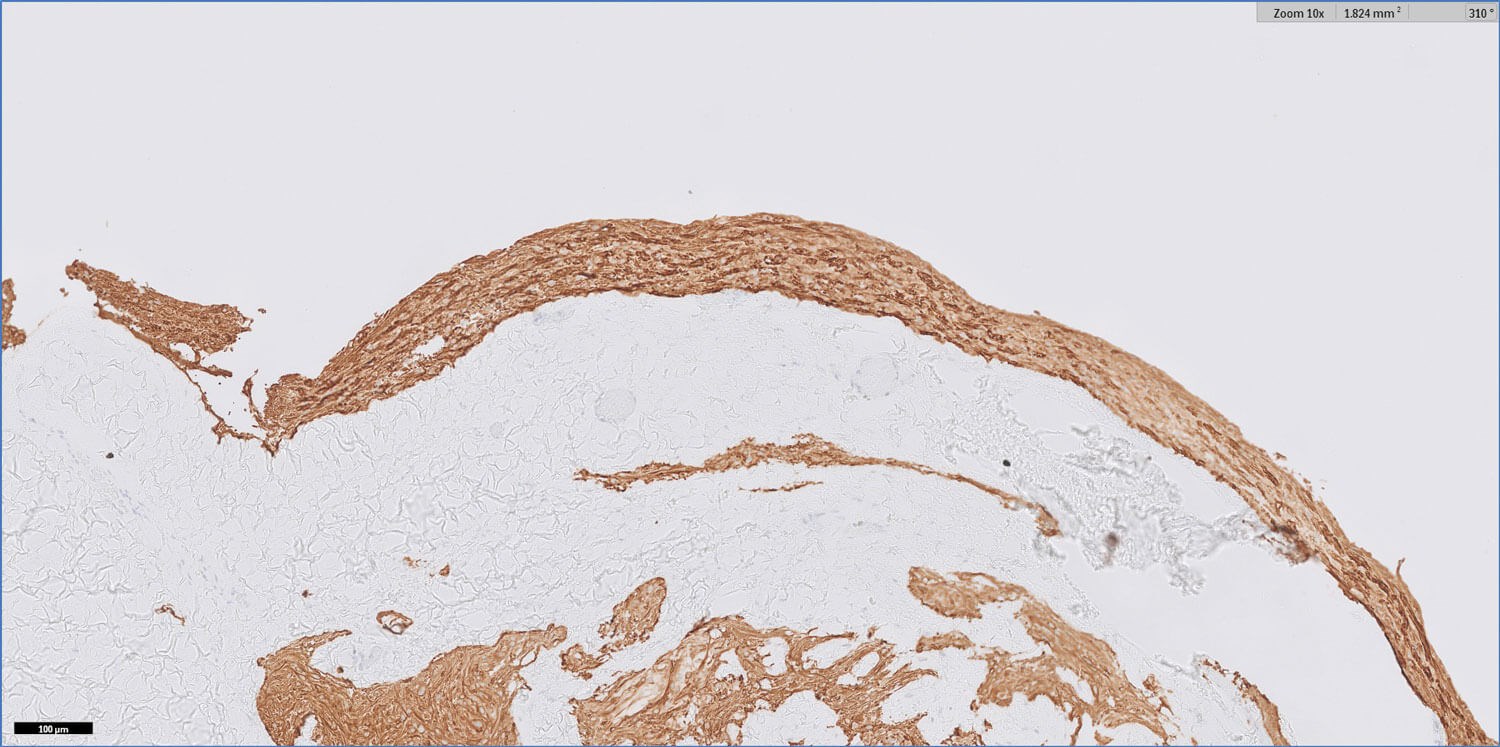
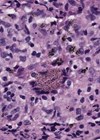
Figure 3: GFAP 10x.
Figures 1–3 show representative H+E-stained sections and glial fibrillary acidic protein (GFAP) immunohistochemistry of the lesion.
- How can these be described?
- Considering the clinicopathological features, what is the diagnosis?
- What are the management options?
Answers
- Figures 1 and 2 show a thick collagenous cyst wall lined by glial cells. Immunohistochemical staining is strongly positive for GFAP (Figure 3). Synaptophysin did highlight the neuroretinal tissue (not shown here). Pancytokeratin (AE1/AE3) was negative.
- The overall features are those of a colobomatous cyst associated with the known clinical history of microphthalmia.
The cyst lining is of disorganised primitive neuroretinal glial tissue, and can show retinal architecture, photoreceptor differentiation or rosette formation. The cyst wall lacks choroidal tissue.
Colobomatous cyst or microphthalmos with cyst is a rare developmental subtype of microphthalmia. It is different from a congenital cystic eye, also known as anophthalmia with cyst, where there is no identifiable globe. Imaging is helpful in these cases. No rudimentary ocular structures would be seen on histopathology.
Microphthalmos with cyst is thought to occur due to failure of the embryonic fissure to close and fuse, and proliferation of neuroectoderm at the persistent fissure. The cyst is present at birth, generally occurs inferonasally or posteriorly, and may produce a bluish mass behind the lower eyelid. Secretions from this neuroectodermal tissue result in cyst formation. Ocular manifestations include: irido- or chorio-retinal coloboma, optic disc coloboma, cloudy cornea, microcornea, shallow anterior chamber or pupillary membrane.
It is usually unilateral, sporadic and without any gender predilection. Bilateral cases are more likely to be familial or syndromic. Systemic abnormalities include microcephalus, corpus callosal agenesis, mid-brain deformities, cardiac conduction defects, cleft lip, saddle nose, pulmonary hypoplasia or renal agenesis. - Management varies, depending on cyst growth and its associated complications, orbit volume and the child’s age. Observation is the management of choice where time is needed for orbital cavity growth and development. Repeated aspiration of the cyst is performed when fluids accumulate and can be curative. Surgical excision of the cyst in-toto is otherwise recommended or complete surgical excision with enucleation of the microphthalmic eye with prosthetic implantation is reserved for patients with no visual potential and complications (such as proptosis, infection or severe ulceration).
COMMENTS ARE WELCOME