Part 1 of this topic can be found here
There are currently no proven cures for inherited retinal disease (IRD). However, multiple avenues of research are being investigated to better understand disease mechanisms and trial potential therapies that may slow or stop disease progression and / or reverse sight loss in IRD [1]. Some of the avenues of research with selected examples will be discussed below.
Supportive management of IRD
Visual rehabilitation
Visual aids such as glasses, contact lenses, magnification aids and specialised computer software can help to optimise residual vision. Photophobia may also be lessened through the use of dark or special filtered glasses or tinted contact lenses – with contact lenses also potentially improving the quality of vision in patients with nystagmus [2].
Treatment of RP-associated cystoid macular oedema
Topical and oral carbonic anhydrase inhibitors have been used in x-linked retinoschisis (XLRS) and for retinitis pigmentosa–associated cystoid macular oedema (RP-CMO) with variable results [3]. Their use may be limited by side-effects, especially for oral agents, such as paraesthesia, fatigue and renal stones. Topical and oral steroids, and non-steroidal anti-inflammatory agents, have also been trialled for RP-CMO with variable effects. The use of intravitreal steroid and anti-vascular endothelial growth factor (anti-VEGF) has been reported in RP-CMO, again, with variable effects that often require re-treatment. A phase II exploratory study is currently underway at Moorfields Eye Hospital, London to assess the efficacy and safety of aflibercept in RP-CMO (AMOUR study, clinicaltrials.gov identifier NCT02661711).
Treatment of vitreous traction / haemorrhage / retinal detachment
Pars plana vitrectomy (PPV) may be required in XLRS-related retinal detachment (RD) / vitreous haemorrhage (VH) without spontaneous resolution in order to avoid amblyopia [3]. It has also been undertaken to remove vitreous traction in XLRS and RP-CMO [3].
Prevention of cell death (retinal neuroprotection)
Inhibition of apoptosis and growth factors
Photoreceptor and / or retinal pigment epithelium (RPE) cell loss by apoptosis, a process of programmed cell death, plays a significant role in IRD. Various retinal disease models have therefore looked at novel ways of directly inhibiting apoptosis [4]. Pharmacological treatment with neurotrophic factors such as nerve growth factor (NGF) has been trialled and shown to promote photoreceptor survival in animal models of RP [5]. A pilot study published earlier this year trialled NGF eye-drops in patients with RP, which demonstrated its safety and possible efficacy [6].
There is evidence from animal studies that ciliary neurotrophic factor (CNTF) delivered either with intravitreal injection of CNTF protein or gene therapy mediated with adeno-associated viral (AAV) vectors exerts a neuroprotective effect on rods, with slowing or halting of retinal degeneration [7]. Recombinant human CNTF has been shown to stimulate cone outer segment regeneration in a rat model of advanced retinal degeneration where rods have already degenerated [8]. In addition, sustained CNTF delivery via implanted devices was shown to preserve cone electroretinogram (ERG) response and function in the same rat model [8]. A phase II/III trial has been undertaken of intravitreal implants of encapsulated human RPE cells engineered to continuously secrete CNTF protein in patients with early (n=68) and late-stage (n=65) RP [9]. Patients were randomly assigned to receive a high- or low-dose implant in one eye and sham surgery in the fellow eye. Primary endpoints were change in best-corrected visual acuity at 12 months for late-stage RP and change in visual field sensitivity at 12 months for early RP.
Neither study showed therapeutic benefit – with some patients experiencing loss of retinal sensitivity that was reversible on removal of the implant. However, a pilot study utilising adaptive optics imaging to investigate in vivo cone structure in three patients with CNTF implants over a 24 month period found that cone density remained stable in eyes with a CNTF implant whereas there was continued cone loss in untreated fellow eyes, suggesting that more sensitive metrics are needed as primary outcome measures in slowly progressive diseases such as RP [10].
Antioxidants
Photoreceptor (PR) cells accumulate reactive oxygen species (ROS) and thereby subject to oxidative stress that is countered by antioxidant defences; with PR loss if the balance is tipped in favour of ROS. Huang et al. (2013) showed that intraperitoneal injection of resveratrol, a potent antioxidant, reduced caspase activation and photoreceptor apoptosis in a rodent retinal detachment model [11]. Lutein, a primary macular pigment, may also offer protection from oxidative damage and supplementation of 12mg/day has been observed to slow loss of mid-peripheral visual field (VF) in non-smoking adults with RP taking vitamin A [12].
Inhibition of necrosis
Photoreceptor loss may also occur from necrosis [4]. Inhibition of a critical mediator of necroptosis, known as receptor interacting protein-1 kinase (RIP1) has been shown to be successful in preventing cone and rod photoreceptor degeneration in a mouse model. RIP3 kinase inhibition has been observed to prevent cone necrosis. Inhibition of the RIP pathway could therefore be a therapeutic target to prevent retinal degeneration, at least in some disease models [13].
Role of inflammation
Inflammation is also thought to play a role in retinal cell death. Intravitreal 17 beta-estradiol has been observed to significantly reduce neuronal apoptosis in a light induced model [14]. Cubilla et al. (2013) found that subcutaneous injections of mifepristone, an antagonist of the glucocorticoid receptor, caused increased apoptotic signals in retinal extracts and apoptosis of the photoreceptors under basal, non-stress conditions [15]; suggesting that glucocorticoids play a critical role in photoreceptor survival.
Other pharmacological therapies
Unoprostone isopropyl is a large-conductance calcium-activated (maxi-K) potassium channel inhibitor currently used in topical form as a treatment for glaucoma. Due to its proposed neuroprotective mechanism and ability to improve macular blood flow, unoprostone isopropyl has been piloted as a potential treatment for patients with RP [16]. However, a recent phase III multicentre trial found no significant difference in the value of mean retinal sensitivity at four central points through HFA (10-2) compared to placebo in patients with RP (clinicaltrials.gov identifier NCT01786395).
Brimonidine, a topical alpha2-agonist, demonstrated neuroprotection by slowing the progression of VF loss in patients with RP in a placebo-controlled, double-masked, randomised study [17]. An exploratory, 12-month, ascending-dose study has also recently been carried out to evaluate the safety and efficacy on visual function following a single injection of a Brimonidine intravitreal implant in one eye of patients with RP (clinicaltrials.gov identifier NCT00661479) – with outcome results pending.
QLT091001 is a synthetic retinoid replacement for 11-cis-retinal that has the potential to improve visual function by restoring this key component of the visual (retinoid) cycle. A phase I study has recently been completed to investigate the safety and visual outcomes of a once-daily oral dose of 40mg/m2/day QLT091001 for seven consecutive days in patients with Leber congenital amaurosis (LCA) or RP due to RPE65 or lecithin:retinol acyltransferase (LRAT) mutations [18]. Eight out of 18 patients (44%) demonstrated at least a 20% increase in functional retinal area on Goldmann visual field testing. Twelve out of 18 (67%) patients demonstrated at least a five ETDRS letter score improvement in one or both eyes [18].
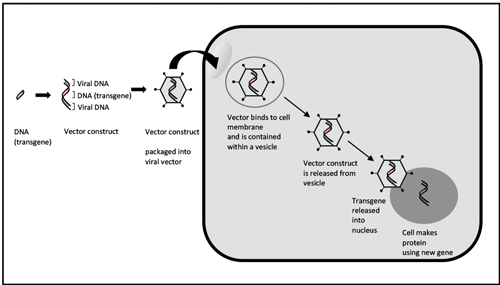
Figure 1: Gene therapy using an adenovirus vector.
Gene therapy
Gene therapy describes a technique whereby a defective gene creating an abnormal or deficient product is replaced with a functioning gene (Figure 1). It is more applicable to the earlier stages of disease before significant retinal deterioration [1]. Common viral vectors used include adeno-associated virus (AAV) and lentivirus.
Ocular administration – The eye is considered an excellent organ for gene therapy. Its immune privilege created by the blood-retina barrier, its small size minimises the amount of vector required, while its easy access and compartmentalisation allows for treatment to be given using intravitreal, intracameral, subretinal or suprachoroidal techniques [19].
Selected active gene therapy trials for IRD – Several gene therapy Phase I/II trials for LCA associated with RPE65 deficiency have shown encouraging preliminary results [19]. A phase III study has recently reported patient’s improvement in ability to navigate a mobility course under different lighting conditions and improvement of full-field light sensitivity threshold testing compared to untreated patients, although visual acuity scores did not significantly improve [20]. A phase I/IIa clinical trial of an optimised vector for RPE65 deficiency has also recently commenced (clinicaltrials.gov identifier NCT02781480).
AAV vectors have been the gene transfer system of choice for human gene therapy, including retinal diseases [21,22]. However, a major limitation is that ABCA4 is larger than the current AAV vector capacity; a challenge that needs to be addressed for other genes that commonly cause inherited retinal disease, including USH2A [23]. Subretinal injection of a lentivirus vector delivering ABCA4 has therefore been developed, given the larger cargo capacity of lentiviruses, and is currently in an on-going Phase I/IIa clinical trial (ClinicalTrials.gov Identifier: NCT01367444). There have been no safety concerns in the first three cohorts of subjects with relatively advanced disease, and no definite evidence of efficacy; with the final cohort with less severe disease now being recruited, with arguably more potential to show benefit [24].
Achromatopsia (ACHM) is considered an excellent target for gene therapy because a significant proportion of viable cone photoreceptors are present [2]. Intravitreal CNTF has been observed to transiently improve cone-mediated function in a CNGB3 mouse model of ACHM [25]. A recent phase I/II clinical study [26] that delivered intravitreal CNTF to ACHM patients with biallelic CNGB3 variants, failed to show any enhancement of cone function, although it has been suggested that the lack of assessment of residual cone number and placement during patient selection may have been a limiting factor in this study [27]. Gene replacement trials have commenced for CNGA3 (clinicaltrials.gov identifier NCT02781480), and anticipated in the near future for CNGB3, both in the UK and USA [2].
Recombinant adeno-associated virus (rAAV)-mediated delivery of the normal RS1 gene to the retina of young knock-out mice has demonstrated long-term retinoschisin expression and rescue of retinal structure and function [3]. There are currently two phase I/II trials assessing the safety and tolerability of two different viral vectors delivered by intravitreal injection in patients with XLRS (clinicaltrials.gov identifier NCT02416622 and NCT02317887) [19].
Figure 2: hESC-derived cone cells. Image provided by Dr Anai Gonzalez Cordero at
The UCL Institute of Ophthalmology, London, UK. Used with permission.
Stem cell therapy
In 2006, Takahashi and Yamanaka’s ground-breaking research demonstrated that induced pluripotent stem cells (iPSCs) could be created by reprogramming mouse fibroblasts, using specific factors, back into an embryonic stem cell-like state [28]. Patient-specific iPSCs have since enabled scientists to investigate specific disease-causing mutations and their associated pathophysiological mechanisms, evaluate novel gene augmentation, gene silencing, and small molecule therapies, and restore function through the transplantation of manufactured cells and tissues [29]. For the reason that iPSCs can be derived directly from adult tissue, the need for embryos can be bypassed. Due to the plasticity of stem cells and their unlimited capacity for self-renewal, however, adverse events such as tumour formation, immune rejection, and the risk of differentiating into unwanted cell types are possible [30]. Studies to date have therefore mainly focused on their safety and tolerability.

Figure 3: hESC-derived RPE cells. Image provided by Dr Anai Gonzalez Cordero at
The UCL Institute of Ophthalmology, London, UK. Used with permission.
A phase I/II, open-Label, multicentre, prospective study was carried out to determine the safety and tolerability of sub-retinal transplantation of human embryonic stem cell (hESC)-derived RPE cells (Figure 3) in patients with Stargardt disease (STGD) and atrophic age-related macular degeneration [30]. No evidence of adverse proliferation, rejection, or serious ocular or systemic safety issues related to the transplanted tissue were found [30]. The adverse events documented were specifically associated with vitreoretinal surgery and immunosuppression [30]. Efficacy data are awaited.
The phase I/IIa, open-Label, prospective study of the safety and tolerability of sub-retinally transplanted human foetal retinal progenitor cells (hRPC), created by ReNeuron, is currently being carried out for patients with RP (clinicaltrials.gov identifier NCT02464436). This follows on from rat model studies where treatment with hRPCs resulted in better visual acuity compared with untreated eyes and greater preservation of the outer nuclear layer on histological analysis [31].
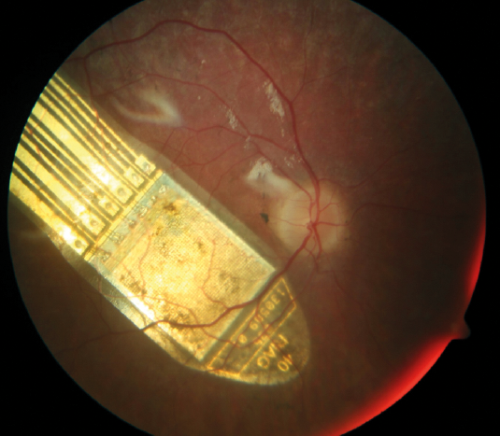
Figure 4: The RETINA IMPLANT Alpha AMS device. Taken from RETINA IMPLANT.
Used with permission. A colour fundus photograph of the right
eye demonstrating the sub-retinal visual implant.
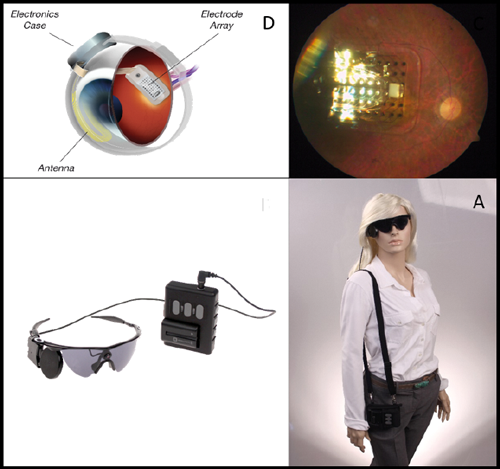
Figure 5: The ARGUS II Retinal Prosthesis System. Taken from Second Sight Medical Products. Used with permission. A = Placement of the electronic implant in and around the eye; B = Fundus photograph demonstrating the implant in-situ; C = A miniature video camera is mounted on a pair of glasses, which sends information to the video processing unit (VPU) via a cable. The VPU processes the information and sends instructions back to the glasses via a cable. These instructions are transmitted wirelessly to an antenna in the retinal implant; D = Model demonstrating how the device is worn.
Artificial vision
The restoration of useful vision can be achieved by bypassing diseased retina and sending signals directly to the brain, or by improving the clarity and magnification of patients’ surroundings. Current devices for patients with RP that are CE marked and available for use in the UK, include [1]:
- The RETINA IMPLANT Alpha AMS is a 3x3mm2 sub-retinal implant with a microchip containing 1600 electrodes that is able to capture light without the use of an external camera, stimulate the retina which delivers signals to the brain (Figure 4). The external handheld device inductively powers the implanted device via an external connecting cable and inductive patch. A clinical trial interim report suggests that sub-retinal implants can restore very-low-vision or low vision in RP patients with light perception vision or worse [32]. The cost of the RETINA IMPLANT Alpha AMS device together with the surgical implantation and visual training / rehabilitation is estimated at $130,000.
- The ARGUS II device (Figure 5) acts by direct stimulation of the relatively preserved inner retina via epiretinal microelectrodes [33]. A small camera mounted on a pair of glasses captures images, which are subsequently converted to a pixelated image by an external video processing unit, and wirelessly transmitted to the retinal implant [33]. The cost of the device with the surgical implantation and visual training / rehabilitation is estimated at $180,000.
Discussion
IRD is the leading cause of blindness certification in the working age population (age 16-64 years) in England and Wales and the second most common in childhood.
Genetic testing is essential in order to further our understanding of IRD and direct appropriate treatments. Whilst diagnostic molecular testing has markedly improved over the last 10 years, approximately 40% of patients tested remain without a molecular diagnosis. We anticipate that continued developments in sequencing performance, technology and interpretation, will increase the number of molecularly proven patients with quicker turnaround times and reduction in cost [34].
Whilst there are currently no proven cures for IRD, much research is being carried out to address all stages of disease, from early to end-stage, with multiple potentially complementary avenues of research being explored, with an ever increasing number of clinical trials anticipated over the next five to ten years.
Gene therapy is best placed for earlier stages of disease where the opportunity still remains to alleviate the underlying defect and prevent further retinal deterioration. More advanced stages of disease would benefit from stem cell therapy and artificial vision [1]. There is no doubt we are entering a new and exciting era of intervention.
References
1. Smith J, Ward D, Michaelides M, et al. New and emerging technologies for the treatment of inherited retinal diseases: a horizon scanning review. Eye (Lond) 2015;29(9):1131-40.
2. Zobor D, Zobor G, Kohl S. Achromatopsia: on the doorstep of a possible therapy. Ophthalmic Res 2015;54(2):103-8.
3. Molday RS, Kellner U, Weber BH. X-linked juvenile retinoschisis: clinical diagnosis, genetic analysis, and molecular mechanisms. Prog Retin Eye Res 2012;31(3):195-212.
4. Chinskey ND, Besirli CG, Zacks DN. Retinal cell death and current strategies in retinal neuroprotection. Curr Opin Ophthalmol 2014;25(3):228-33.
5. Lambiase A, Aloe L. Nerve growth factor delays retinal degeneration in C3H mice. Graefes Arch Clin Exp Ophthalmol 1996;234 Suppl1:S96-100.
6. Falsini B, Iarossi G, Chiaretti A, et al. NGF eye-drops topical administration in patients with retinitis pigmentosa, a pilot study. J Transl Med 2016;14:8.
7. Wen R, Tao W, Li Y, Sieving PA. CNTF and retina. Prog Retin Eye Res 2012;31(2):136-51.
8. Li Y, Tao W, Luo L, et al. CNTF induces regeneration of cone outer segments in a rat model of retinal degeneration. PloS one 2010;5(3):e9495.
9. Birch DG, Weleber RG, Duncan JL, et al. Randomized trial of ciliary neurotrophic factor delivered by encapsulated cell intraocular implants for retinitis pigmentosa. Am J Ophthalmol 2013;156(2):283-92.e1.
10. Talcott KE, Ratnam K, Sundquist SM, et al. Longitudinal study of cone photoreceptors during retinal degeneration and in response to ciliary neurotrophic factor treatment. Invest Ophthalmol Vis Sci 2011;52(5):2219-26.
11. Huang W, Li G, Qiu J, et al. Protective effects of resveratrol in experimental retinal detachment. PloS one 2013;8(9):e75735.
12. Berson EL, Rosner B, Sandberg MA, et al. Clinical trial of lutein in patients with retinitis pigmentosa receiving vitamin A. Arch Ophthalmol 2010;128(4):403-11.
13. Sato K, Li S, Gordon WC, et al. Receptor interacting protein kinase-mediated necrosis contributes to cone and rod photoreceptor degeneration in the retina lacking interphotoreceptor retinoid-binding protein. J Neurosci 2013;33(44):17458-68.
14. Mo MS, Li HB, Wang BY, et al. PI3K/Akt and NF-kappaB activation following intravitreal administration of 17beta-estradiol: neuroprotection of the rat retina from light-induced apoptosis. Neuroscience 2013;228:1-12.
15. Cubilla MA, Bermudez V, Marquioni Ramella MD, et al. Mifepristone, a blocker of glucocorticoid receptors, promotes photoreceptor death. Invest Ophthalmol Vis Sci 2013;54(1):313-22.
16. Akiyama M, Ikeda Y, Yoshida N, et al. Therapeutic efficacy of topical unoprostone isopropyl in retinitis pigmentosa. Acta Ophthalmol 2014;92(3):e229-34.
17. Merin S, Obolensky A, Farber MD, Chowers I. A pilot study of topical treatment with an alpha2-agonist in patients with retinal dystrophies. J Ocul Pharmacol Ther 2008;24(1):80-6.
18. Scholl HP, Moore AT, Koenekoop RK, et al. Safety and Proof-of-Concept Study of Oral QLT091001 in retinitis pigmentosa due to inherited deficiencies of retinal pigment epithelial 65 Protein (RPE65) or Lecithin: Retinol Acyltransferase (LRAT). PloS one 2015;10(12):e0143846.
19. Garoon RB, Stout JT. Update on ocular gene therapy and advances in treatment of inherited retinal diseases and exudative macular degeneration. Curr Opin Ophthalmol 2016;27(3):268-73.
20. Schimmer J, Breazzano S. Investor outlook: significance of the positive LCA2 gene therapy phase III results. Hum Gene Ther Clin Dev 2015;26(4):208-10.
21. Warrington KH Jr. Herzog RW. Treatment of human disease by adeno-associated viral gene transfer. Human Genetics 2006;119(6):571-603.
22. Allocca M, Doria M, Petrillo M, et al. Serotype-dependent packaging of large genes in adeno-associated viral vectors results in effective gene delivery in mice. J Clin Invest 2008;118(5):1955-64.
23. Han Z, Conley SM, Naash MI. Gene therapy for Stargardt disease associated with ABCA4 gene. Adv Exp Med Biol 2014;801:719-24.
24. Weleber RG, Stout T, Lauer AK, et al. Early findings in a Phase I/IIa clinical program for Stargardt disease (STGD1, MIM #248200). Invest Ophthlamol Vis Sci 2015;56(7):3819.
25. Marangoni D, Vijayasarathy C, Bush RA, et al. Intravitreal ciliary neurotrophic factor transiently improves cone-mediated function in a CNGB3-/- mouse model of achromatopsia. Invest Ophthalmol Vis Sci 2015;56(11):6810-22.
26. Zein WM, Jeffrey BG, Wiley HE, et al. CNGB3-achromatopsia clinical trial with CNTF: diminished rod pathway responses with no evidence of improvement in cone function. Invest Ophthalmol Vis Sci 2014;55(10):6301-8.
27. Langlo C, Dubis A, Michaelides M, Carroll J. CNGB3-Achromatopsia clinical trial with CNTF: diminished rod pathway responses with no evidence of improvement in cone function. Invest Ophthalmol Vis Sci 2015;56(3):1505.
28. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006;126(4):663-76.
29. Giacalone JC, Wiley LA, Burnight ER, et al. Concise review: patient-specific stem cells to interrogate inherited eye disease. Stem Cells Transl Med 2016;5(2):132-40.
30. Schwartz SD, Regillo CD, Lam BL, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet 2015;385(9967):509-16.
31. Luo J, Baranov P, Patel S, et al. Human retinal progenitor cell transplantation preserves vision. J Biol Chem 2014;289(10):6362-71.
32. Stingl K, Bartz-Schmidt KU, Besch D, Chee CK, Cottriall CL, Gekeler F, et al. Subretinal Visual Implant Alpha IMS--Clinical trial interim report. Vision research. 2015;111(Pt B):149-60
33. Luo YH, da Cruz L. The Argus((R)) II Retinal Prosthesis System. Prog Retin Eye Res 2016;50:89-107.
34. Neveling K, Collin RW, Gilissen C, et al. Next-generation genetic testing for retinitis pigmentosa. Hum Mutat 2012;33(6):963-72.
Declaration of competing interests:
This work was supported by grants from the National Institute for Health Research Biomedical Research Centre at Moorfields Eye Hospital National Health Service Foundation Trust and UCL Institute of Ophthalmology, Fight For Sight (UK), The Macular Society (UK), Moorfields Eye Hospital Special Trustees, Moorfields Eye Charity, by a multiuser equipment grant from The Wellcome Trust [099173/Z/12/Z], the Foundation Fighting Blindness (FFB; USA), Bayer UK, and Retinitis Pigmentosa Fighting Blindness. Professor Michel Michaelides is a recipient of an FFB Career Development Award.
COMMENTS ARE WELCOME




