Part 2 of this topic can be found here
Inherited Retinal Disease (IRD) is the leading cause of blindness certification in the working age population (age 16-64 years) in England and Wales and the second most common in childhood [1]. The term IRD incorporates a large group of disorders that are clinically and genetically heterogeneous [2].
IRDs may be sub-categorised in several ways including: (i) according to photoreceptor involvement, for example, rod or cone; when both are involved the nomenclature reflects the order in which the photoreceptors are affected, for example, rod-cone dystrophies affect the rods prior to the cones. Also by (ii) natural history – stationary or progressive disorders; (iii) isolated macular or generalised retinal involvement; and (iv) isolated ocular or additional systemic manifestations (syndromic) [3].
IRDs may be inherited in an autosomal dominant (AD), autosomal recessive (AR) or X-linked (XL) manner. IRDs are genetically diverse and complex. While certain phenotypes can be caused by disease-causing sequence variants in different genes, mutations within the same gene can result in varying phenotypes [4], with marked inter- and intra-familial variability being commonplace [3]. This, together with the large number of genes that have been identified in IRDs (>300), can result in significant diagnostic challenge.
Well-characterised IRDs and those subject to / anticipated to be subject to clinical trial will be described below.
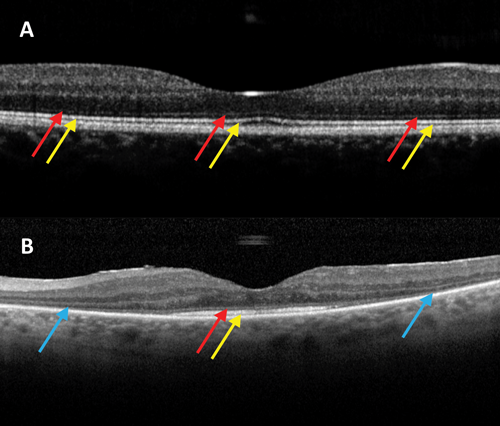
Figure 1: OCT images centered on the macula in a patient without RP (A) and a patient with RP (B). Red arrows: intact external limiting membrane; Yellow arrows: intact ellipsoid zone (IS/OS junction); Blue arrows: loss of peripheral macular outer retinal layers, with relative preservation centrally. Used with permission.
Retinitis pigmentosa
Retinitis pigmentosa (RP) represents a group of disorders which are the most common IRD, with an incidence of approximately one in 3000 in the USA and Europe [3,5]. RP is a clinically variable rod-cone dystrophy, characterised by nyctalopia, and progressive visual field loss initially, with central visual involvement later in the disease process (Figure 1). Clinical examination is characterised by bone-spicule retinal pigmentation, generalised vascular attenuation and optic disc pallor.
RP is a genetically heterogeneous disorder with over 60 genes currently known to cause RP [6,7]. The most common inheritance pattern is autosomal recessive (AR; 30-40%), with mutations in the gene USH2A being the most common cause [8]. AD inheritance occurs in 20-30% of RP, with mutations in the Rhodopsin (RHO) gene being the most common [8]. XL RP occurs in 15-20%, with mutations in the RP GTPase regulator (RPGR) gene accounting for the vast majority [8]. There is no known family history in 40-50% of patients at presentation [9]. XLRP and ARRP are the most severe forms of RP with onset often in childhood. ADRP is milder and often of later onset. Digenic RP is rare and is caused by disease-causing variants occurring in both PRPH2 and ROM1 [8].
RP can be isolated (most common) or syndromic. Examples of syndromic RP include: Usher syndrome (RP and neurosensory hearing loss), Bardet Biedl syndrome (RP with obesity, developmental delay, polydactyly, hypogonadism and renal abnormalities), Kearns-Sayre syndrome (RP with external ophthalmoplegia, ataxia and heart block), Abetalipoproteinemia (RP with fat malabsorption and progressive ataxia) and Refsum disease (RP with anosmia, deafness, ataxia, neuropathy and icthyosis) [10]. The latter two syndromes can be managed with dietary modification.
Multiple avenues of research are being explored in RP including gene and stem cell therapy, pharmacological approaches and artificial vision, with on-going or anticipated clinical trials [11-13].
Leber congenital amaurosis
Leber congenital amaurosis (LCA) is a severe congenital / early-onset IRD with an incidence of three per 100,000 births [14]. The inheritance pattern is AR with 22 genes identified to date which account for approximately 70-80% of cases [15]. Three of the most common causative genes are: GUCY2D (6-21%), CEP290 (10-20%) and RPE65 (5-16%) [16].
Patients present at birth or early infancy with severe visual impairment, nystagmus and amaurotic pupils [5,16]. Other symptoms and signs may include photoattraction or photophobia, nyctalopia, the oculodigital sign, keratoconus and cataract [16]. LCA can be isolated (most common) or syndromic, e.g. Joubert and Senior-Loken syndrome. Fundus examination ranges from a normal appearance especially at presentation, to marked maculopathy, white retinal dots and bone-spicule pigment migration [16].
Electroretinography is typically non-detectable or severely reduced in LCA [16]; and is helpful in distinguishing between other causes of congenital / early-onset nystagmus and reduced visual behaviour, including achromatopsia, congenital stationary night blindness and albinism.
Gene therapy-based clinical trials are on-going or planned in the near future for several genetic forms of LCA including RPE65, MERTK, CEP290, AIPL1 and GUCY2D [13,17,18].
Stargardt disease
Stargardt disease (STGD) is the most common inherited macular dystrophy, with a prevalence of one in 10,000 [19], and is caused by recessive sequence variants in the ATP-binding cassette transporter (ABCA4) gene. STGD is characterised by toxic accumulation of lipofuscin and related products including the bis-retinoid N-retinylidene-N-retinylethanolamine (A2E) within retinal pigment epithelium (RPE) cells, resulting in RPE cell dysfunction / death, with subsequent photoreceptor cell dysfunction / loss.

Figure 2: Fundus autofluorescence imaging in a patient with Stargardt disease. Red arrow: increased and decreased autofluorescence in areas where flecks are present; Yellow arrow: reduced autofluorescence at the central macula; Blue arrow: surrounding area of increased signal.
Over 900 disease-causing sequence variants have been identified in ABCA4 to date, resulting in marked phenotypic heterogeneity including variable age of onset and severity [19,20]. Typical presentation occurs during the first or second decade of life with bilateral central visual loss and dyschromatopsia. Earlier onset (childhood-onset STGD) is associated with a worse prognosis and more severe ABCA4 sequence variants, compared to later onset (adulthood-onset STGD and foveal-sparing STGD) [13]. Fundus appearance may be normal in the early stages, with development of increasing macular atrophy / bull’s-eye maculopathy, and yellow-white flecks at the level of the RPE which are associated with characteristic areas of increased and decreased autofluorescence (Figure 2), which changes over time. Spectral-domain optical coherence tomography (SD-OCT) shows loss of outer retinal architecture at the central macula [21].
Electrophysiological assessment is useful for both diagnosis and, moreover, informing advice on prognosis: Group 1 patients have a normal full-field ERG (ffERG) in the presence of severe macular dysfunction (pattern ERG or multifocal ERG); Group 2 patients have generalised cone system dysfunction in addition to macular dysfunction; and Group 3 patients have both generalised cone and rod system dysfunction in addition to macular dysfunction [22,23]. Group 1 is associated with a better prognosis compared to Group 3; with Group 3 associated with a greater rate of progression, worse visual acuity and loss of peripheral vision over time [22].
Multiple avenues of intervention are currently being explored, including gene therapy, stem cell therapy and pharmacological approaches, with on-going clinical trials in all three areas [13,21].
Achromatopsia
Achromatopsia (ACHM) is an autosomal recessive cone dysfunction syndrome [24]. Disease-causing sequence variants have been identified in five genes that encode components of the cone-specific phototransduction cascade (CNGA3, CNGB3, GNAT2, PDE6C and PDE6H) [25-30], and also ATF6, which has a role in the maintenance of the endoplasmic reticulum and cellular homeostasis [31]. CNGA3 and CNGB3 account for approximately 70-80% of patients with ACHM [24].
ACHM has a prevalence of one in 30,000, with presentation at birth or early infancy with poor central vision, pendular nystagmus, marked photophobia / photoaversion and reduced or absent colour vision [32,33]. Patients prefer mesopic conditions and have normal night vision. Two forms exist – complete and incomplete – patients with incomplete ACHM retain residual cone function and thereby have residual colour vision and mildly better VA [33,34]. ACHM may be progressive, however, the changes that occur are slow and subtle in most patients, and does not correlate with age or genotype [35].

Figure 3: OCT image centered on the macula taken from a patient with achromatopsia. This image demonstrates the presence of a foveal hyporeflective zone.
The fundus appearance in ACHM is often normal but RPE disturbance or atrophy at the macula can be seen [33]. Electrophysiological assessment demonstrates reduced, or absent cone responses with normal rod responses. SD-OCT findings are variable in ACHM and have implications for anticipated gene therapy trials; outer retinal architecture may be classified as: (1) continuous inner segment ellipsoid (ISe), (2) ISe disruption, (3) absent ISe, (4) foveal hyporeflective zone (Figure 3), and (5) outer retinal atrophy [36].
Successful rescue of multiple small and large animal models of CNGA3 and CNGB3 associated ACHM have led to on-going and anticipated gene replacement clinical trials for both forms of ACHM [13,35].
X-linked retinoschisis
X-linked retinoschisis (XLRS) affects 1:5,000-1:20,000 [37], and is caused by disease-causing sequence variants in the gene RS1, with over 200 variants identified to date. The encoded protein, retinoschisin, is secreted and binds to both photoreceptors and bipolar cells to help maintain the integrity of the retina [32]. While penetrance is complete, clinical expression is highly variable.

Figure 4: OCT image centred on the macula taken from a patient with x-linked retinoschisis. A: spokewheel-like maculopathy. B: typical macular schisis as seen at the level of the green line observed in A.
Boys typically present in the first or second decade of life with strabismus, amblyopia, anisometropia, or having failed school vision screening [32,38]. The characteristic fundus abnormality is a cystic spokewheel-like maculopathy (foveal schisis), present in about two-thirds of males, and most readily observed with SD-OCT (Figure 4). Fifty percent of affected males show additional peripheral retinal changes – often a (usually lower temporal) peripheral retinoschisis [19]. Other peripheral retinal changes include pigmentary retinopathy, perivascular sheathing and capillary closure. Retinal detachment and vitreous haemorrhage may complicate X-linked retinoschisis [32].
Full-field ERG shows a reduced b- to a-wave ratio, ‘electronegative ERG’, suggestive of inner retinal dysfunction. Carrier females have a normal fundus appearance and normal EOG and ERG [19].
The visual prognosis is variable but relatively good in childhood as long as retinal detachment or vitreous haemorrhage does not occur. Central vision may deteriorate slowly in adulthood with development of macular atrophy in the fourth to sixth decade [19].
Successful structural and functional rescue of small animal models of XLRS have led to two on-going gene replacement clinical trials using an intravitreal route of administration compared to subretinal delivery in other IRD gene therapy trials (Clinicaltrials.gov NCT02317887 and NCT02416622).
Assessment of IRD
1. Structural assessment of IRD
Imaging techniques such as SD-OCT, fundus autofluorescence (FAF) imaging and adaptive optics scanning laser ophthalmoscopy (AOSLO) provide enhanced assessment of retinal architecture and have proven invaluable in the diagnosis and monitoring of IRD [21]. These techniques allow for earlier detection of changes in retinal structure and more sensitive monitoring of progression than clinical examination alone.
SD-OCT is a non-invasive rapid technique allowing qualitative and quantitative analysis of retinal lamination. It has allowed the ready detection of disease in childhood where delay in diagnosis is common due to the often subtle signs on clinical examination in early stages – including childhood-onset STGD and XLRS. External limiting membrane thickening prior to the development of outer retinal loss in STGD has been detected in children as young as five years of age using SD-OCT [39]. Measurement of ISe width or area has also been shown to be a reliable and reproducible measure of progression in RP and will likely be used as a metric in planned clinical trials [11].
FAF imaging uses the autofluorescent properties of lipofuscin and related metabolites to detect their presence within RPE cells [21]. Increased autofluorescence represents RPE cells with greater amounts of lipofuscin, whereas reduced autofluorescence represents lower levels of RPE-containing lipofuscin and / or RPE/photoreceptor cell loss or atrophy. Characteristic FAF patterns can be seen in several IRDs, which aid diagnosis including STGD and PRPH2-associated retinopathy. FAF imaging can also be useful in monitoring progression either by accurately detecting atrophy enlargement over time, e.g. STGD [21], or documenting the change in size of perimacular rings of increased signal that can be seen in, for example, RP [11].
AOSLO is a high-resolution non-invasive technique that makes use of scanning laser ophthalmoscopy to visualise the photoreceptor and RPE mosaics in vivo. The ability to directly visualise the cells being targeted with novel therapies including gene replacement is transformational and will allow improved determination of participant suitability and selection for intervention, and thereby clinical trial design and the likelihood of detecting safety and efficacy signals robustly and sensitively [11,21,24].
2. Functional assessment of IRD
Visual field testing (VFT) is a non-invasive technique used to detect central and peripheral visual field (VF) defects, thus contributing to the diagnosis and monitoring of IRD. Perimetry employs the use of stimuli of varying size and luminance to test specific areas of the visual field and can be presented in a static or kinetic fashion. Using a systematic approach, these tests can be repeated to look for progression of IRD over time.
Microperimetry (MP) makes use of central visual field testing whilst simultaneously observing each point of retinal stimulation [40]. In this way, it is possible to compare retinal morphology with visual function. There is evidence that retinal sensitivity in the macula correlates well with outer retinal thickness in RP [40]. This, together with literature suggesting that retinal sensitivity is related to quality of life [41], supports the use of MP in the evaluation of IRD.
The ‘Hill-of-Vision’ (HoV) is a function of retinal sensitivity based on information acquired from VFT and MP. Visual Field Modelling and Analysis (VFMA) is an innovative software currently being used to create detailed HoV maps that allow for a more thorough assessment by using all available retinal sensitivity data, and thereby arguably more sensitive and reliable measure of change over time which will be valuable both for IRD clinical practice and clinical trials [42].
3. Molecular assessment of IRD
Inheritance patterns may be determined by careful construction of a pedigree; however, molecular genetic testing can provide confirmation of mode of inheritance together with identification of the specific underlying causative gene and sequence variant(s). This is particularly useful when clinical diagnosis is uncertain, for example, in conditions with marked phenotypic variability. It also aids better informed genetic counselling, advice on prognosis and potential participation in studies and clinical trials.
Single gene testing is useful for conditions with strong phenotype and genotype correlations, for example, XLRS and STGD [43]. Next-generation sequencing (NGS) allows for large numbers of genes to be sequenced simultaneously. This technique is employed for large gene panel testing, which is more appropriate for conditions such as RP where a large variety of genes can be responsible [43,44]. Currently multi-gene panel testing and directed single gene screening enables a molecular diagnosis to be confidently made in 50-60% of IRD cases [3]. Higher detection rates are possible with research-based methodologies such as whole exome and whole genome sequencing.
References
1. Liew G, Michaelides M, Bunce C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16-64 years), 1999-2000 with 2009-2010. BMJ Open 2014;4(2):e004015.
2. Smith J, Ward D, Michaelides M, et al. New and emerging technologies for the treatment of inherited retinal diseases: a horizon scanning review. Eye (Lond) 2015;29(9):1131-40.
3. Chiang JP, Lamey T, McLaren T, et al. Progress and prospects of next-generation sequencing testing for inherited retinal dystrophy. Expert Rev Mol Diagn 2015;15(10):1269-75.
4. Ran X, Cai WJ, Huang XF, et al. ‘RetinoGenetics’: a comprehensive mutation database for genes related to inherited retinal degeneration. Database: the journal of biological databases and curation. 2014.
5. Francis PJ. Genetics of inherited retinal disease. J R Soc Med 2006;99(4):189-91.
6. RetNet, the Retinal Information Network:
http://www.sph.uth.tmc.edu/RetNet/
7. Nash BM, Wright DC, Grigg JR, et al. Retinal dystrophies, genomic applications in diagnosis and prospects for therapy. Transl Pediatr 2015;4(2):139-63.
8. The Association for Research in Vision and Ophthalmology (ARVO) annual meeting. Fort Lauderdale, Florida, USA. Abstracts. Invest Ophthalmol Vis Sci 1998;39(4):S1-1134.
9. Openshaw A, Branham K, Heckenlively J. Understanding Retinitis Pigmentosa. Michigan: University of Michigan, Kellogg Eye Center; 2008.
10. Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis 2006;1:40.
11. Tee JJ, Smith AJ, Hardcastle AJ, Michaelides M. RPGR-associated retinopathy: clinical features, molecular genetics, animal models and therapeutic options. Br J Ophthalmol 2016;100(8):1022-7.
12. RP Fighting Blindness. Research 2016.
http://www.rpfightingblindness.org.uk/
index.php?tln=research
Last accessed August 2016.
13. ClinicalTrials.gov
https://clinicaltrials.gov/
Last accessed August 2016.
14. Koenekoop RK. An overview of Leber congenital amaurosis: a model to understand human retinal development. Surv Ophthalmol 2004;49(4):379-98.
15. Fahim AT, Daiger SP, Weleber RG. Retinitis Pigmentosa Overview. GeneReviews. University of Washington: Seattle; 1993.
16. Chacon-Camacho OF, Zenteno JC. Review and update on the molecular basis of Leber congenital amaurosis. World J Clin Cases 2015;3(2):112-24.
17. Sundaram V, Moore AT, Ali RR, Bainbridge JW. Retinal dystrophies and gene therapy. Eur J Pediatr 2012;171(5):757-65.
18. Aboshiha J, Dubis AM, van der Spuy J, et al. Preserved outer retina in AIPL1 Leber’s congenital amaurosis: implications for gene therapy. Ophthalmology 2015;122(4):862-4.
19. Michaelides M, Hunt DM, Moore AT. The genetics of inherited macular dystrophies. J Med Genet 2003;40(9):641-50.
20. Zernant J, Xie YA, Ayuso C, et al. Analysis of the ABCA4 genomic locus in Stargardt disease. Hum Mol Genet 2014;23(25):6797-806.
21. Tanna P, Strauss RW, Fujinami K, Michaelides M. Stargardt disease: clinical features, molecular genetics, animal models and therapeutic options. Br J Ophthalmol 2016 [Epub ahead of print].
22. Lois N, Holder GE, Bunce C, et al. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch Ophthalmol 2001;119(3):359-69.
23. Fujinami K, Lois N, Davidson AE, et al. A longitudinal study of stargardt disease: clinical and electrophysiologic assessment, progression, and genotype correlations. Am J Ophthalmol 2013;155(6):1075-88.
24. Aboshiha J, Dubis AM, Carroll J, et al. The cone dysfunction syndromes. Br J Ophthalmol 2016;100(1):115-21.
25. Kohl S, Hamel C. Clinical utility gene card for: Achromatopsia – update 2013. Eur J Hum Genet 2013;21(11).
26. Thiadens AA, Slingerland NW, Roosing S, et al. Genetic etiology and clinical consequences of complete and incomplete achromatopsia. Ophthalmology 2009;116(10):1984-9.
27. Thiadens AA, Roosing S, Collin RW, et al. Comprehensive analysis of the achromatopsia genes CNGA3 and CNGB3 in progressive cone dystrophy. Ophthalmology 2010;117(4):825-30.
28. Kohl S, Baumann B, Rosenberg T, et al. Mutations in the cone photoreceptor G-protein alpha-subunit gene GNAT2 in patients with achromatopsia. Am J Hum Genet 2002;71(2):422-5.
29. Thiadens AA, den Hollander AI, Roosing S, et al. Homozygosity mapping reveals PDE6C mutations in patients with early-onset cone photoreceptor disorders. Am J Hum Genet 2009;85(2):240-7.
30. Kohl S, Coppieters F, Meire F, et al. A nonsense mutation in PDE6H causes autosomal-recessive incomplete achromatopsia. Am J Hum Genet 2012;91(3):527-32.
31. Kohl S, Zobor D, Chiang WC, et al. Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nature Genet 2015;47(7):757-65.
32. Molday RS, Kellner U, Weber BH. X-linked juvenile retinoschisis: clinical diagnosis, genetic analysis, and molecular mechanisms. Prog Retin Eye Res 2012;31(3):195-212.
33. Zobor D, Zobor G, Kohl S. Achromatopsia: on the doorstep of a possible therapy. Ophthalmic Res 2015;54(2):103-8.
34. Pokorny J, Smith VC, Pinckers AJ, Cozijnsen M. Classification of complete and incomplete autosomal recessive achromatopsia. Graefes Arch Clin Exp Ophthalmol 1982;219(3):121-30.
35. Aboshiha J, Dubis AM, Cowing J, et al. A prospective longitudinal study of retinal structure and function in achromatopsia. Invest Ophthalmol Vis Sci 2014;55(9):5733-43.
36. Sundaram V, Wilde C, Aboshiha J, et al. Retinal structure and function in achromatopsia: implications for gene therapy. Ophthalmology 2014;121(1):234-45.
37. George ND, Yates JR, Moore AT. X linked retinoschisis. Br J Ophthalmol 1995;79(7):697-702.
38. Sikkink SK, Biswas S, Parry NR. X-linked retinoschisis: an update. J Med Genet 2007;44(4):225-32.
39. Lee W, Noupuu K, Oll M, et al. The external limiting membrane in early-onset Stargardt disease. Invest Ophthalmol Vis Sci 2014;55(10):6139-49.
40. Battu R, Khanna A, Hegde B, et al. Correlation of structure and function of the macula in patients with retinitis pigmentosa. Eye (Lond) 2015;29(7):895-901.
41. Sugawara T, Sato E, Baba T, et al. Relationship between vision-related quality of life and microperimetry-determined macular sensitivity in patients with retinitis pigmentosa. Jpn J Ophthalmol 2011;55(6):643-6.
42. Weleber RG, Smith TB, Peters D, et al. VFMA: Topographic analysis of sensitivity data from full-field static perimetry. Transl Vis Sci Technol 2015;4(2):14.
43. Chiang JP, Trzupek K. The current status of molecular diagnosis of inherited retinal dystrophies. Curr Opin Ophthalmol 2015;26(5):346-51.
44. Khan KN, Chana R, Ali N, et al. Advanced diagnostic genetic testing in inherited retinal disease: experience from a single tertiary referral centre in the UK National Health Service. Clin Genet 2016 [Epub ahead of print].
Take home message
-
IRD is the leading cause of blindness certification in the working age population (age 16-64 years) in England and Wales and the second most common in childhood.
-
RP is the most common form of IRD overall, while Stargardt is the most common macular dystrophy.
-
Diagnostic molecular testing is increasingly available, with identification of the disease-causing gene in approximately 60% of patients.
Declaration of competing interests:
This work was supported by grants from the National Institute for Health Research Biomedical Research Centre at Moorfields Eye Hospital National Health Service Foundation Trust and UCL Institute of Ophthalmology, Fight For Sight (UK), The Macular Society (UK), Moorfields Eye Hospital Special Trustees, Moorfields Eye Charity, by a multiuser equipment grant from The Wellcome Trust [099173/Z/12/Z], the Foundation Fighting Blindness (FFB; USA), Bayer UK, and Retinitis Pigmentosa Fighting Blindness. Professor Michel Michaelides is a recipient of an FFB Career Development Award.
COMMENTS ARE WELCOME



