The authors describe the process set up in Manchester for the optimum delivery and assessment of a new gene therapy treatment for patients with RPE65 IRD.
Inherited retinal dystrophies (IRDs) are the second commonest cause of severe visual impairment in childhood and the most frequent in the working age population [1], and there are several hundred known responsible genes. The uptake of modern genetic testing has meant that the majority of patients with IRD are able to have a timely and accurate molecular diagnosis, informing prognosis for vision and allowing genetic counselling.
Gene therapy was approved for the first time in the UK in 2019 for one of the most severe IRD subtypes, that caused by RPE65 mutations. This new treatment offers hope for patients and families with IRD but also presents new challenges for the clinician in terms of patient selection, managing expectations, surgical techniques and understanding of the effect of the gene therapy treatment.
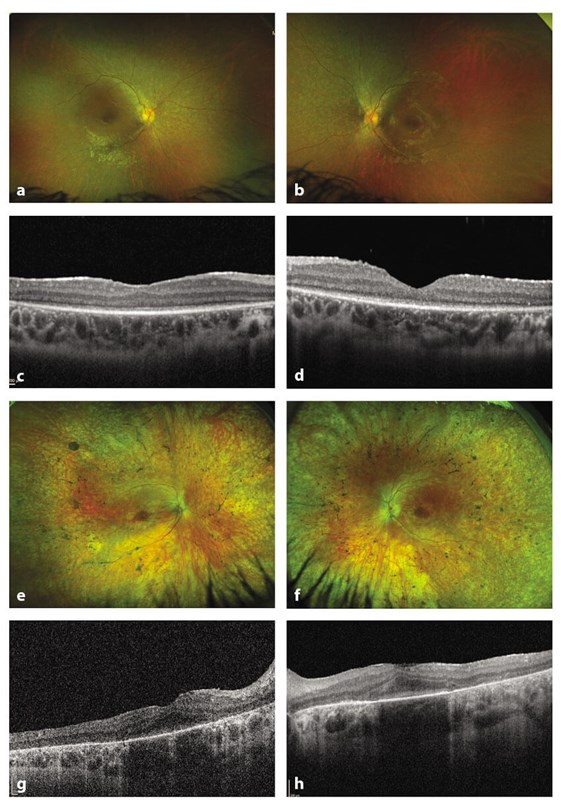
Figure 1: Optos wide field digital imaging and autofluoresence
in a child and adult with RPE65 inherited retinal disease.
a-d: 11-year-old child with RPE65 IRD showing relative preservation of retinal structure.
e-h: 30-year-old female with retinal pigmentary changes and disruption of outer retinal layers on OCT.
Autosomal recessively inherited mutations in RPE65 cause Leber’s congenital amaurosis / early onset severe rod-cone dystrophy, severe early childhood-onset retinal dystrophy and juvenile retinitis pigmentosa (Figure 1). The RPE65 protein is expressed in the retinal pigment epithelium (RPE) and is involved in the visual cycle [2]. The typical phenotype of this condition is an early onset retinal dystrophy with some preservation of cone function but early loss of rod function, resulting in severe night blindness. A visual acuity of 6/30-6/60 may be achieved with some stability in the first decade [3,4]. Electroretinography may show some residual cone activity, whilst rod activity is usually extinguished. Optical coherence tomography scanning may also demonstrate some preservation of central macular thickness and structure surrounded by areas of retinal thinning [5]. Over time, progressive reduction in visual acuity, particularly after the age of 20 years, results in severe visual impairment accompanied by progressive loss of visual field. The relative preservation in childhood of retinal structure and modest visual function opens up the possibility of restoring function by gene therapy in this subtype of IRD.
Gene therapy for RPE-65 IRD is delivered by subretinal injections of modified adeno-associated virus (AAV) with cDNA of RPE65 [6,7]. A randomised controlled phase III study of AAV2-hRPE65-v2 (Voretigene nepavovec) carried out in 31 subjects aged three years and older showed that treated patients had a rapid improvement in their performance of a standardised multi-luminance mobility test (MLMT); a test of their ability to navigate a course in different luminance levels that was sustained through to one year, and which was significantly different to controls. A secondary outcome measure of full-field light sensitivity threshold testing (FST), which measures the lowest illumination seen over the entire retinal field in the dark adapted eye, showed a rapid greater than 2 log unit improvement that also remained stable over one year (whilst no improvement was seen in the control group). There was also some evidence of improvement in visual acuity, Goldman visual fields and macular sensitivity in the treated group [8]. Overall, these studies provided some reassurance of the safety and tolerability of subretinal injection of Voretigene, and hope of some potential stabilisation or improvement in visual function.
The clinical trials data for Voretigene gene therapy for RPE65 IRD have been evaluated by the National Institute for Health and Care Excellence (NICE) and the improvement in mobility and functional vision, even though it was a small change, was considered important for affected patients. As a result, approval for the use of Voretigene nepavovec (Luxterna®) was granted in September 2019 as the first gene therapy treatment for IRD (https://www.nice.org.uk/guidance/hst11/resources/voretigene-neparvovec-for-treating-inherited-retinal-dystrophies-caused-by-rpe65-gene-mutations-pdf-50216253809605). In the UK, there are three centres approved for gene therapy treatment for patients with RPE65 IRD (Oxford, London and Manchester), and this article describes the process set up in Manchester (by the Manchester Ocular Gene Therapy Group) for the optimum delivery and assessment of this new treatment.
Identification of suitable patients: the multidisciplinary process
The identification of suitable patients is the key first stage of instigation of gene therapy treatment. Genomic Testing for IRD is delivered by the North West Genomic Laboratory Hub to serve the populations of the North West (including Manchester and Liverpool), North East (including Leeds, Newcastle and Sheffield) and the East of England (including Nottingham, Cambridge and Leicester). The North West laboratory as a result has a database of over 8000 patients, having carried out genetic testing for IRD including RPE65 since 2006. All patients with RPE65 likely causal variants were identified from the database, and the referring clinicians across the North of England informed of the new Luxterna treatment and invited to refer their patients. Patient information sheets and a bespoke referral form was developed to support new referrals for treatment.
An inclusive multidisciplinary team (MDT) was set up between Manchester Centre for Genomic Medicine and Manchester Royal Eye Hospital to facilitate collaborative working between vitreo-retinal surgeons, paediatric ophthalmologists, geneticists, clinical scientists, ophthalmic imaging, nursing and genetic counsellors. MDT meetings are convened bimonthly to discuss genetics (whether the genetic RPE65 variant identified in the patient meets the criteria for pathogenicity) and ophthalmology (if the patient in addition meets the clinical criteria for treatment such as visual function, macular thickness) and assess eligibility for treatment for the patients who have been referred. At the same time, patient knowledge, expectations and consent are discussed, and a pre-treatment / post-treatment follow-up plan is put in place.
Pre-treatment screening and assessment of visual function
Once a patient with RPE65 IRD has been discussed in the MDT and confirmed to be suitable for treatment from a genetic perspective, they then need to fulfil three clinical criteria before the treatment can go ahead. The patient needs to have:
- Three or more-disc areas without atrophy or pigmentary degeneration within the posterior pole
- OCT showing more than a 100‑micrometre thickness in an area of the retina within the posterior pole
- A remaining visual field (VF) within 30⁰ of fixation.
Pre and post-treatment imaging, electrophysiology and psychophysics are therefore crucial to assess patient eligibility for treatment, and also assess the impact of treatment on visual function, particularly when any treatment benefit is unlikely to be measured by visual acuity alone. Preoperative imaging includes wide field retinal imaging including colour and auto fluorescence (to assess posterior pole degeneration), and Heidelberg Optical Coherence Tomography (to assess retinal thickness). Ganzfeld and Multifocal electroretinograms are conducted using ISCEV standards, and visual fields are conducted using static 10-2 macular threshold perimetry. Contrast sensitivity and colour vision are tested. Low luminance visual acuity is tested using three levels of illumination down to 0.34cd.m-2, and dark-adapted retinal sensitivity is tested using FST testing. It may not be possible for a patient to complete all tests depending on age and cooperation. Imaging and OCT is carried out at each postoperative visit, and where possible, psychometric testing preoperatively, at three to six months postoperatively and after one year.
The patient journey
Once a patient has been identified as being suitable from genetic and clinical perspectives, then treatment planning with the family can begin. A key aspect of this has been the management of expectations ensuring that patients receive a balanced understanding of possible and likely outcomes. Time has been set aside to give families an opportunity to ask about the surgery (with vitreoretinal surgeons, paediatric ophthalmologists and genetic counsellors as appropriate); patients are generally seen first with their families in the genetic retinal clinic or paediatric ophthalmology clinic to discuss the availability of treatment and understand their views. While most have been known to clinical genetics for some years, a number have been either newly diagnosed or referred from outside the region. There remain uncertainties around long-term outcomes of this treatment, and while many have so far been keen to proceed with treatment in the hope that it will reduce further deterioration and / or to stabilise vision, others have declined, keen to gather more information and confidence in the treatment before proceeding.
The surgical procedure for delivery of gene therapy
Meticulous planning of the surgical procedure is important, as the patient needs to be treated preoperatively with oral steroids, and ordering, storage and preparation of the Voretigene drug needs to be arranged. If both eyes meet the criteria for treatment, then the eye with worse visual acuity is generally treated first. The surgical delivery of the gene is usually performed under general anaesthesia since most eligible patients are either children or young adults, and the procedure takes approximately one hour of surgical time. Patients are started on oral steroids (1mg/kg;maximum 40mg) three days prior to the surgery with a gradual taper after seven days for a total duration of 17 days.
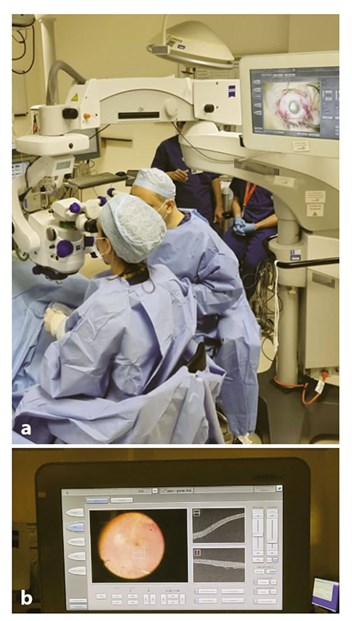
Figure 2: Surgery for gene therapy treatment
(a) with use of the intraoperative OCT
(b) to visualise the subretinal injection of Voretigene.
On the day of the surgical procedure, the drug has to be thawed and prepared in aseptic conditions. This can take approximately 60 minutes, and so this has to be taken into account in scheduling the operating list. A pars plana vitrectomy (PPV) is carried out, followed by the subretinal injection of 0.3ml of Voretigene neparvovec at the posterior pole. An intraoperative optical coherence tomography (OCT) is useful in ascertaining the delivery of the drug at the subretinal location (Figure 2). Fluid / air exchange is then performed and the eye is left air-filled at the end of the procedure. Patients are instructed to posture supine for 24 hours, in order to minimise the risk of macular folds due to the large volume of subretinal injection and to avoid displacing the gene outside the inferior arcade. Patients are usually kept in overnight to facilitate the posturing regimen.
The risks of the surgery relate to both the PPV and the subretinal gene delivery itself. These include risk of endophthalmitis, retinal detachment, high and low intraocular pressure, and the risks of immune response towards the treatment (which is why oral steroids are given), risk of macular damage (such as development of a macular hole or fold linked to macular detachment and the volume of injected drug).
Although the clinical trials advocated treating the second eye within 14 days from the first (to potentially reduce the immune response), many clinicians, including those of the Manchester Ocular Gene Therapy Group, decide on the timing between treatments on an individual basis, depending on the outcome of the first eye, patient preference, as well as other factors. In clinical practice, we have usually treated the second eye around three to six months after the first surgery.
Our experience so far suggests that the majority of treated patients experience an increase in retinal sensitivity measured by FST, and improvement in low luminance navigational vision, with little if any improvement of the central visual acuity. There is not enough data on the longevity of this effect, but early results are very promising, and offer an opportunity for increasing role of gene therapy in other inherited retinal diseases.
Current and future gene therapy for inherited retinal disease
Reconfiguration of genomic testing services now supports access to genomic diagnosis for all patients with IRD across the UK, so that identification of patients with RPE65 IRD should be achievable at an earlier stage of the condition. Suitable patients may now wish to consider gene therapy in the hope that this will at least stabilise their visual function. The next few years will provide further information on the effectiveness of Voretigene gene therapy for patients with RPE65 IRD, as well as opportunities for treatment of other IRD subtypes.
References
1. Bunce C, Zekite A, Wormald R, Bowman R. Is there evidence that the yearly numbers of children newly certified with sight impairment in England and Wales has increased between 1999/2000 and 2014/2015? A cross-sectional study. BMJ Open 2017;7:e016888.
2. Xue L, Gollapalli DR, Maiti P, et al. A palmitoylation switch mechanism in the regulation of the visual cycle. Cell 2004;117(6):761-71.
3. Lorenz B, Gyurus P, Preising M, et al. Early-onset severe rod cone dystrophy in young children with RPE65 mutations. Invest Ophthalmol Vis Sci 2000;41(9):2735-42.
4. Paunescu K, Wabbels B, Preising MN, Lorenz B. Longitudinal and cross-sectional study of patients with early-onset severe retinal dystrophy associated with RPE65 mutations. Graefes Arch Clin Exp Ophthalmol 2005;243(5):417-26.
5. Chung eDC, Bertelsen M, Lorenz B, et al. The Natural History of Inherited Retinal Dystrophy Due to Biallelic Mutations in the RPE65 Gene. Am J Ophthalmol 2019;199:58-70.
6. Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med 2008;358:2231-9.
7. Maguire AM, High KA, Auricchio A, et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet 2009;374(9701):1597-605.
8. Russell S, Bennet J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 2017;390:849-60.
Declaration of competing interests: None declared.
COMMENTS ARE WELCOME





